Diseases Related to the Sorting of Prohormones
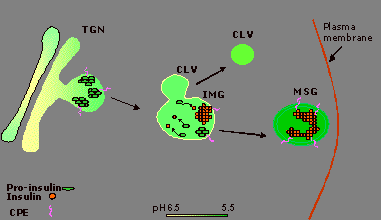
The focus of research in our laboratory lies primarily with characterizing the mechanisms of protein trafficking to the regulated secretory pathway in endocrine cells. Specifically, we have identified the membrane form of carboxypeptidase E (CPE) as a sorting receptor for several prohormones, including pro-opiomelanocortin (POMC), proenkephalin and p roinsulin. We have developed a model for the trafficking of proinsulin through the regulated secretory pathway (Figure 1). Hexamers of p roinsulin bind to the sorting receptor, CPE, at the trans-Golgi network, or TGN. Within the immature secretory granule, p roinsulin is converted to insulin through the sequential actions of the prohormone convertases, PC1 and PC2. Mature insulin is then retained in the immature secretory granule, and is eventually stored within the mature secretory granule. We are making use of this model in order to determine a cellular mechanism underlying a form of hyperp roinsulinemia.
Familial hyperp roinsulinemia (FH) is a genetic condition resulting in the presence of significantly elevated levels of plasma p roinsulin. Some patients develop diabetes as a result of the increased secretory demand. Associated with this disease are a number of point mutations in the p roinsulin gene: three that result in a biologically defective form of insulin, and four (His10Asp, Arg65Leu, Arg65Pro and Arg65His) that result in the defective sorting and processing of p roinsulin. Studies from animal and cell models have shown that the sorting of His10Asp (also known as B10) is inefficient, in that it is partially diverted to the constitutive secretory pathway. However, the sorting of the other p roinsulin mutants has not been studied. We hypothesize that the mutant p roinsulins found in FH are missorted to the constitutive secretory pathway, due to impaired binding to CPE either at the level of the TGN or within the immature secretory granule. The inefficient sorting and secretion of the mutant p roinsulins would then account for the high plasma p roinsulin levels observed in FH. In order to study the sorting behavior of the mutant p roinsulins found in FH, we are engineering an endocrine cell line to produce mutant forms of p roinsulin. We make use of biochemical and cell biological techniques, such as radioimmunoassay, high pressure liquid chromatography, fluorescence immunocytochemistry and receptor binding assays, in order to examine the trafficking of the mutant p roinsulins. The results from these experiments suggest that the mutant forms of p roinsulin found in FH show some impairment in sorting to the regulated secretory pathway and in binding to CPE.
The Leu34Phe ProCART Mutation causes CART Deficiency leading to Obesity in Humans
Cocaine and amphetamine regulated transcript (CART) is an anorexigenic neuropeptide, and its absence in CART / mice results in obesity. We show that obese humans bearing a Leu34Phe mutation in pro-CART have diminished serum levels of bioactive CART, and elevated amounts of partially processed pro-CART. Leu34Phe pro-CART expressed in AtT20 cells was missorted and secreted constitutively as unprocessed proCART; while wild type pro-CART was sorted to the regulated secretory pathway vesicles and processed to CART. The defective intracellular sorting of Leu34Phe proCART would account for the reduced levels of bioactive CART in affected humans. These results suggest that the obesity observed in humans bearing the Leu34Phe mutation could be due to a putative deficiency in hypothalamic bioactive CART.
 BACK TO TOP
BACK TO TOP