Secretory Granule Biogenesis
Secretory granules (or large dense-core vesicles, LDCVs) are unique organelle in which neuropeptides and/or hormones are packaged and stored for secretion via the regulated secretory pathway (RSP) upon stimulation in neuroendocrine and endocrine cells. Secretory granule formation is therefore a prerequisite step to fulfill the precise physiological functions of neuropeptides and hormones.
Despite vigorous studies to understand the mechanism(s) for secretory granule biogenesis for the past several decades, not much information is available so far.
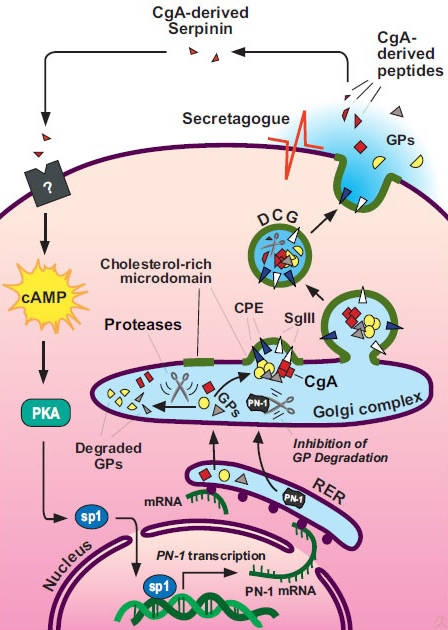
One of the goals in our laboratory is to uncover the molecular mechanism(s) responsible for the biogenesis of secretory granules in neuroendocrine and endocrine cells. In order to achieve this goal, we employ techniques such as electron microscopy, confocal microscopy as well as other biochemical and cell biological methods in these studies to analyze secretory granule biogenesis. We utilize neuroendocrine and endocrine model cell lines such as PC12, AtT-20 and 6T3 cells. In addition, we utilize technologies to knock-down gene expression (i.e. antisense RNA, siRNA) in neuroendocrine cells such as PC12 to study the role of proteins involved in dense-core secretory granule biogenesis. Using an antisense RNA technology, we have identified that chromogranin A (CgA) is a key molecule involved in secretory granule biogenesis in neuroendocrine PC12 cells, and re-expression of CgA rescued the RSP and hormone secretion in endocrine 6T3 cells (Kim et al., 2001). In vivo study using transgenic mice expressing antisense RNAs against CgA has confirmed the role of CgA in secretory granule biogenesis in adrenal chromaffin cells (Kim et al., 2005). Moreover, using DNA microarray technology, real-time PCR and proteomics, we identified another key protein, protease nexin-1 (PN-1), involved in secretory granule biogenesis (Kim and Loh, 2006). PN-1 was up-regulated in the presence of CgA in 6T3 cells and prevented the degradation of granule proteins in the Golgi complex (Kim and Loh, 2006). Currently we are interested in a signal transduction pathway for PN-1 induced by CgA in AtT-20 and 6T3 endocrine cells.
Trafficking, processing and induction of granule biogenesis
Cholesterol and secretory granules
Cholesterol is an abundant lipid in eukaryotic membranes, implicated in numerous structural and functional capacities. We have investigated the mechanism by which cholesterol affects secretory granule biogenesis in vivo using Dhcr7-/- and Sc5d-/- mouse models of the human diseases, Smith-Lemli-Opitz syndrome (SLOS) and lathosterolosis. These homozygous-recessive multiplemalformation disorders are characterized by the functional absence of one of the last two enzymes in the cholesterol biosynthetic pathway, resulting in the accumulation of precursors. Cholesterol-deficient mice exhibit a significant decrease in the numbers of secretory granules in the pancreas, pituitary and adrenal glands. Moreover, there was an increase in morphologically aberrant granules in the exocrine pancreas of Dhcr7-/- acinar cells. Regulated secretory pathway function was also severely diminished in these cells, but could be restored with exogenous cholesterol. Sterol precursors incorporated in artificial membranes resulted in decreased bending rigidity and intrinsic curvature compared with cholesterol, thus providing a cholesterol-mediated mechanism for normal granule budding, and an explanation for granule malformation in SLOS and lathosterolosis.
 BACK TO TOP
BACK TO TOP