Mice expressing reduced levels of hepatic G6Pase-α activity do not develop age-related insulin resistance or obesity.
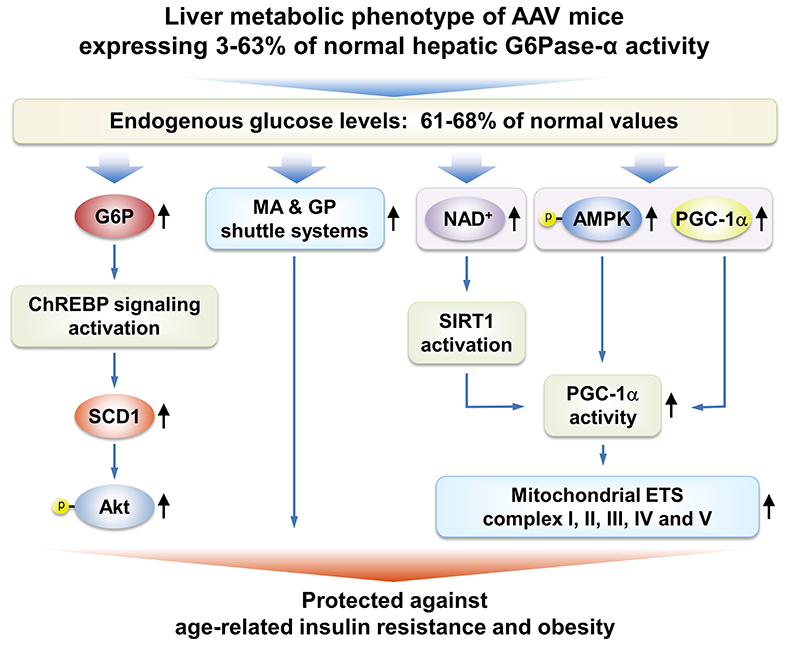
We have shown that gene therapy mediated by rAAV8-G6PC, a recombinant adeno-associated virus pseudotype 2/8 (rAAV8) vector expressing human G6Pase-alpha directed by the human G6PC promoter/enhancer, normalizes blood glucose homeostasis in G6pc knockout (G6pc−/−) mice for 70–90 weeks. The treated G6pc−/− mice, which express 3–63% of normal hepatic G6Pase-alpha activity (AAV mice), produce endogenous hepatic glucose at 61–68% levels of wild-type littermates, have a leaner phenotype, and exhibit fasting blood insulin levels more typical of young adult mice. We showed that, unlike wild-type mice, the lean AAV mice have elevated caloric intake but do not develop age-related obesity or insulin resistance. Pathway analysis showed that signaling by hepatic ChREBP, a glucose-activated transcription factor that improves glucose tolerance and insulin signaling, is activated in AAV mice. In addition, several longevity factors in the calorie restriction pathway, including the NADH shuttle systems, NAD+ concentrations, and the AMPK/sirtuin1/PGC-1alpha pathway, are up-regulated in the livers of AAV mice. Our study suggests that full restoration of normal G6Pase-alpha activity by gene therapy will not be required to confer significant therapeutic benefits in GSD-Ia. The finding that a moderate reduction of hepatic G6Pase-alpha activity in mice may promote a leaner phenotype and prevent the development of age-related decline in insulin sensitivity lends weight to the suggestion that G6Pase-alpha constitutes a potential pharmaceutical target for treating type 2 diabetes and provides insight into the safety margin in such activity-reducing therapies.
Click image to view.
Several signaling pathways in the livers of AAV mice contribute to the beneficial metabolic phenotype. The ChREBP-SCD1-Akt signaling pathway, activated by elevated hepatic G6P levels, leads to improved insulin signaling and glucose tolerance. Increases in the expression of the malate-aspartate (MA) and glycerol-3-phosphate (GP) shuttle systems, which couple mitochondrial ATP synthesis to the regeneration of NAD+, confer insulin sensitivity and resistance to age-related obesity. Activation of the AMPK/SIRT/PGC-1α pathway, which leads to increased expression of mitochondrial electron transport chain complexes and protects against age-related mitochondrial dysfunction, has three components: (1) increased expression of PGC-1α, a master regulator of mitochondrial biogenesis; (2) PGC-1α activation via deacetylation by SIRT1, which is activated by elevated cellular concentrations of NAD+; and (3) PGC-1α activation via phosphorylation by p-AMPK.
Molecular mechanisms of neutrophil dysfunction in GSD-Ib
The underlying cause of GSD-Ib neutropenia is an enhanced neutrophil apoptosis, but patients also manifest neutrophil dysfunction of unknown etiology. Previously, we show G6PT interacts with the enzyme G6Pase-beta to regulate the availability of G6P/glucose in neutrophils. A deficiency in G6Pase-β activity in neutrophils impairs both their energy homeostasis and functionality. We showed that G6PT–deficient neutrophils from human GSD-Ib patients are similarly impaired, an energy impairment that is characterized by reduced glucose uptake and diminished levels of intracellular G6P, lactate, ATP, and NADPH, while functional impairment is reflected in lowering of neutrophil respiratory burst, of chemotaxis, and of calcium mobilization. We also showed that the expression and membrane translocation of the NADPH oxidase subunit p47phox is downregulated in G6PT–deficient neutrophils, explaining why respiratory burst activity is impaired. We further showed that the HIF-1alpha/PPAR-gamma pathway, which directly impacts neutrophil respiratory burst, chemotaxis, and calcium mobilization, is activated in G6PT–deficient neutrophils. We also found that exposing human G6PT–deficient neutrophils to a PPAR-gamma antagonist improves their function. Taken together, our results demonstrate that the underlying cause of neutrophil dysfunction in GSD-Ib arises from impaired neutrophil energy homeostasis and activation of the HIF-1alpha/PPAR-gamma pathway. The insight into the etiology of neutrophil dysfunction in GSD-Ib should facilitate the development of novel therapies for this disorder.
Functional analysis of mutations in GSD-Irs
The enzyme G6Pase-beta is embedded in the ER membrane and catalyzes the hydrolysis of G6P to glucose and phosphate. To date, 33 separate G6Pase-beta mutations have been identified in GSD-Irs patients, but only the p.R253H and p.G260R missense mutations have been characterized functionally for pathogenicity. We functionally characterized 16 of the 19 known missense mutations, using a sensitive assay based on a recombinant adenoviral vector–mediated expression system to demonstrate pathogenicity. Twelve missense mutations completely abolish G6Pase-beta enzymatic activity while the p.M116V, p.T118R, p.S139I, and p.R189Q mutations retain 1.1%, 1.3%, 49%, and 45%, respectively, of wild-type G6Pase-beta activity. A database of residual enzymatic activity retained by the G6Pase-beta mutations will serve as a reference for evaluating genotype-phenotype relationships.
Long-term safety and efficacy of liver-directed gene therapy in murine GSD-Ia
GSD-Ia is characterized by impaired glucose homeostasis and the long-term complication of hepatocellular adenoma (HCA,) which may undergo malignant transformation to hepatocellular carcinoma (HCC). In a long-term study, we showed that 70–90 week-old rAAV-G6PC–treated G6pc−/− mice expressing 3% or greater of normal hepatic G6Pase-α activity maintain glucose homeostasis and show no evidence of HCA/HCC. Despite the encouraging results, to translate rAAV–mediated gene therapy in murine GSD-Ia to the clinic, the safety concerns associated with administering large doses of the rAAV vector must be addressed. We therefore generated rAAV-co-G6PC, a rAAV8 vector expressing a codon-optimized (co) human G6Pase-alpha (co-G6PC) directed by the human G6PC promoter/enhancer, and investigated the impact of codon optimization strategies on translation efficiency. In a 66–80-week study, we showed that the rAAV-co-G6PC vector is more efficacious than the rAAV-G6PC vector in directing hepatic G6Pase-alpha expression. As expected, the treated mice expressing 3% or more of normal hepatic G6Pase-α activity display normal metabolic profiles, maintain normoglycemia over a 24-hour fast, show no evidence of HCA/HCC, and are protected against age-related obesity and insulin resistance. Among the twenty-six 66–80-week-old rAAV8-G6PC- and rAAV8-co-G6PC–treated G6pc−/− mice, twelve expressed hepatic G6Pase-alpha activity 3% or more of normal hepatic G6Pase-α activity. One mouse expressing 0.9% of normal hepatic G6Pase-alpha activity developed HCA and two mice expressing 0.9% and 1.3% of normal hepatic G6Pase-alpha activity, respectively developed HCC, establishing the threshold of transgene expression required to avoid HCA/HCC. We further showed that ChREBP signaling, which improves glucose tolerance and insulin sensitivity, is activated in all rAAV–treated mice but that activation of ChREBP signaling is significantly attenuated in the HCC nodules.
Liver-directed gene therapy for murine GSD-Ib
We showed that systemic administration of rAAV-CBA-G6PT, a rAAV8 vector expressing human G6PT directed by the chicken b-actin (CBA) promoter/CMV enhancer, delivered the G6PT transgene to the liver and normalized metabolic abnormalities in murine GSD-Ib. However, the five transduced GSD-Ib mice that lived to age 52–70 weeks expressed less than 4% of wild-type hepatic G6PT activity and two mice developed HCA, with one undergoing malignant transformation. Studies have shown that the choice of transgene promoter not only impacts targeting efficiency and tissue-specific expression, but also the level of immune response or tolerance to the therapy. We therefore examined the safety and efficacy of rAAV8-G6PT, a rAAV8 vector expressing human G6PT directed by the tissue-specific human G6PC promoter/enhancer. Among the fifteen 60–78 week-old rAAV–treated G6pt−/− mice expressing 2–62% of wild-type hepatic G6PT activity, only one mouse expressing 6% of normal hepatic G6PT activity developed HCA. The rAAV–treated mice, including the HCA–bearing mouse, displayed normal hepatic fat storage, normal glucose tolerance profiles, and maintained normoglycemia over a 24-hour fast. The rAAV–treated mice also exhibit a leaner phenotype and are protected against age-related insulin resistance. We further showed that activation of hepatic ChREBP signaling, which improves glucose tolerance and insulin sensitivity, is one mechanism that protects the rAAV-GPE-G6PT–treated G6pt−/− mice against age-related obesity and insulin resistance.
 BACK TO TOP
BACK TO TOP