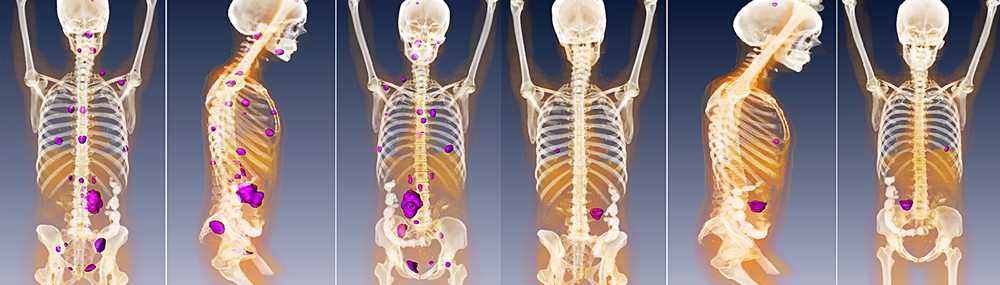
The success of pheochromocytoma treatment depends upon several factors; the most important include the presence of metastasis, genetics, location, and overall extent of the disease. Malignant pheochromocytoma can only be determined by the presence of metastasis or tumor spreading (tumors in locations such as the bones, liver, lungs, or lymph nodes).
The only curative treatment for pheochromocytoma is the complete surgical removal of the tumor. The long-term prognosis of patients after resection of a single sporadic pheochromocytoma is excellent.
Even after tumor removal, there is still a risk that pheochromocytoma or paraganglioma might return (called recurrence). Recurrence is more likely for those who have paraganglioma, those who are younger, those whose family members also have the disease, and those who had large tumors.1
There is currently no cure for malignant pheochromocytoma. Radiotherapy, or the use of high-powered X-rays or radioactive beams to destroy tumors, can assist in shrinking some malignant tumors.2 Tumor shrinkage can lessen the clinical signs and symptoms of pheochromocytoma by reducing the production of hormones and in some cases may allow for surgery.
Although not curative, medications are used to control the clinical signs and symptoms of both benign and malignant pheochromocytoma.
 BACK TO TOP
BACK TO TOP