Research
The incredible diversity and heterogeneity of interneurons was observed over a century ago, with Ramon y Cajal hypothesizing in Recollections of My Life that "The functional superiority of the human brain is intimately linked up with the prodigious abundance and unaccustomed wealth of the so-called neurons with short axons." Although interneurons constitute the minority (20%) of neurons in the brain, they are the primary source of inhibition and are critical components in the modulation and refinement of the flow of information throughout the nervous system. Abnormal development and function of interneurons has been linked to the pathobiology of numerous neurodevelopmental & psychiatric disorders such as epilepsy, schizophrenia and autism spectrum disorders. Interneurons are an extremely heterogeneous cell population, with distinct transcriptomes, morphologies, neurochemical markers electrophysiological properties. With the advent of new technologies such as single cell sequencing and spatial transcriptomics to dissect gene expression and connectivity patterns, the classification of interneurons into specific subtypes is continuously evolving.
GABAergic interneurons are born in the ventral forebrain during embryogenesis and undergo a prolonged migratory period to populate nearly every brain region. However, our understanding of the developmental mechanisms that generate this diversity of GABAergic cell types remains poorly characterized. The goal of my lab is to characterize the transcriptomic and epigenetic mechanisms that drive interneuron fate decisions during normal development and in disease models. We take a multifaceted approach, utilizing both in vitro and in vivo strategies to identify candidate mechanisms that regulate interneuron fate decisions. We strive to develop cutting-edge techniques that will overcome the many challenges faced when studying interneuron development. We believe that our pursuits will act as a springboard for future research and provide continue generating new insights into forebrain GABAergic interneuron development.
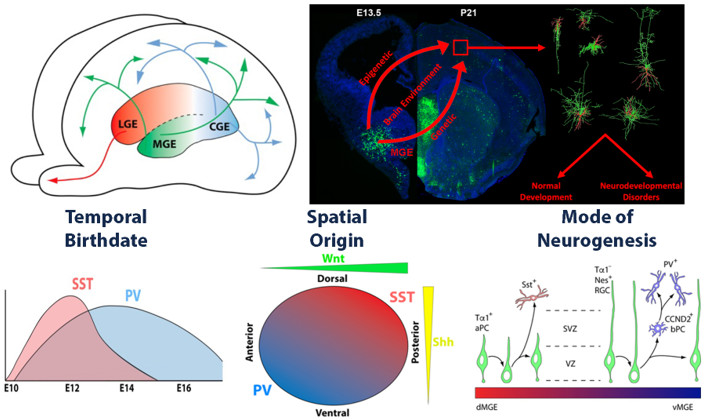
Generating an ‘Epigenome Atlas’ of the embryonic mouse forebrain
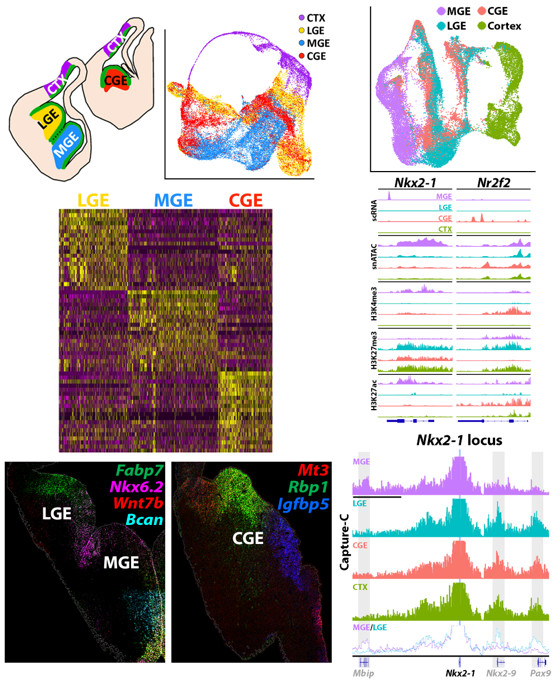
Genome wide association studies (GWAS) indicate that >90% of disease-associated single nucleotide polymorphisms (SNPs) are located outside of coding regions, and many neurological disorders have been linked directly to SNPs in enhancer regions. Thus, a thorough characterization of the epigenomic landscape during neurogenesis is critical to understand normal brain development and potential disease etiologies. To this end, we characterized the transcriptome (scRNA-seq), chromatin accessibility (snATAC-seq), histone modifications to identify active promoters & enhancers (CUT&Tag), and higher order chromatin structure (Hi-C) in 4 regions of the embryonic mouse brain that give rise to inhibitory (MGE, LGE & CGE) and excitatory (cortex) neurons. These findings were recently published in a series of studies (Lee…Petros eLife, 2022 ![]() ; Rhodes…Petros Nature Communications, 2022
; Rhodes…Petros Nature Communications, 2022 ![]() ; Lee…Petros STAR Protocols, 2022
; Lee…Petros STAR Protocols, 2022 ![]() ). And this entire ‘Epigenome Atlas’s dataset is publicly available in an easily searchable platform on the UCSC Genome Browser that can be found here.
). And this entire ‘Epigenome Atlas’s dataset is publicly available in an easily searchable platform on the UCSC Genome Browser that can be found here.
To highlight some of the most important findings from these studies: (1) A prevailing theory in the literature is that cycling neural progenitors are relatively homogeneous and cell fate markers don’t emerge until cells become postmitotic. Using a Nestin-GFP mouse, we identified significant transcriptional heterogeneity in VZ cells, with many genes specifically enriched in RGCs in the MGE, LGE or CGE. We verified many of these expression patterns via fluorescent in situ hybridization (FISH) and found that some genes were enriched in specific subdomains (e.g., dorsal vs. ventral) within each GE. This observation indicates that VZ cells already express spatially restricted genes that pattern the GE and likely are critical for future cell fate decisions. (2) Our snATAC-seq & histone modification data revealed significant differences in chromatin accessibility between brain regions. We identified candidate brain region specific promoter-enhancer interactions, many of which were supported by H3K4me3 and H3K27ac data. (3) Our Hi-C data revealed brain region-specific chromatin interactions as critical fate-determining genes. For example, the transcription factor Nkx2.1 is expressed in the MGE and is critical for all MGE-derived cells. There is a direct interaction between the Nkx2.1 promoter and the Mbip locus in the MGE that is not present in the LGE, CGE or cortex. Conversely, in these non-MGE regions, the Nkx2.1 promoter interacts directly with the Nkx2.9 and Pax9 locus, whereas this interaction is not observed in the MGE. We are currently exploring how perturbation of these interactions at the Nkx2.1 locus affect gene expression and cell fate. To our knowledge, this was the first study to explore differential epigenetic landscape in distinct regions of the embryonic mouse brain, and it highlights the importance of characterizing multiple epigenetic mechanisms in vivo because many of these differences would not have been observed using in vitro cell cultures.
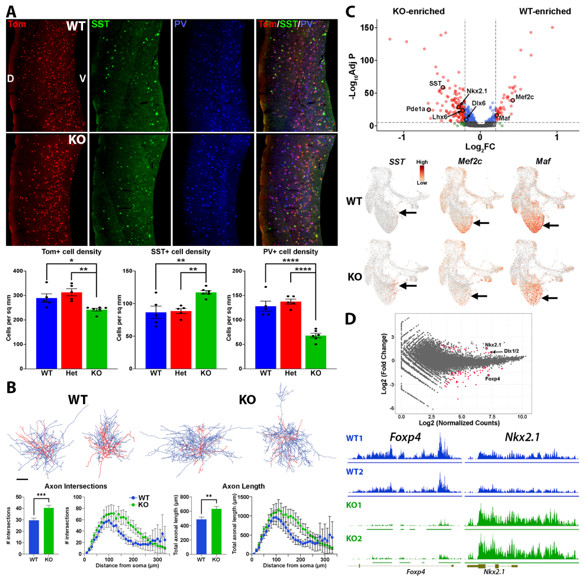
Perturbation of epigenetic processes alters gene expression, interneuron fate & function, and ultimately animal behavior
Now that we established a ‘ground truth’ for gene expression and chromatin organization in the embryonic mouse brain, our lab is well positioned to explore how perturbation of specific genes and epigenetic processes affect interneuron fate and maturation. We strive for a multifaceted approach, assessing changes in mutant mice at a genetic, molecular, cellular and behavioral level to truly understand the function of a gene or epigenetic process in neurodevelopment. While numerous studies are ongoing, two of our recent studies are highlighted below.
There are ~300 human genes coding for proteins that drive epigenetic modifications (readers, writers and erasers of epigenetic marks, chromatin remodelers, etc.), and these genes are highly intolerant to genetic variation. Predictably, heterozygous loss-of-function variants in these genes leads to numerous neurodevelopmental diseases (NDDs). In a recent publication, we characterized the role of Enhancer of zeste homolog 2 (Ezh2) in development of MGE-derived interneurons (Rhodes…Petros Front Cell Neuro 2024 ![]() ). Ezh2 is the primary methyltransferase targeting trimethylation of histone 3 lysine 27 (H3K27me3), which is critical for gene repression. In humans, EZH2 variants lead to Weaver Syndrome, a complex disease with varying degrees of intellectual disabilities. Loss of Ezh2 in mice can lead to premature neuronal differentiation, migration defects and changes in neuronal fate, but the role of Ezh2 in forebrain interneurons has not been explored. We demonstrated that loss of Ezh2 in the MGE reduced the overall number of MGE-derived interneurons in the cortex and caused a shift in fate with an increase in SST+ and a decrease in PV+ interneurons. Consistent with this result, single cell sequencing of the MGE revealed increased expression of SST-related genes cells in the KO MGE, whereas genes predictive of PV-fated interneurons were decreased. We found no changes in the intrinsic e-phys properties of PV+ or SST+ cells in the KO cortex. However, PV+ cells in the KO displayed more complex axonal arbors, with increased axonal length and arborization compared to WT PV+ cells. Despite a global reduction of H3K27me3 in Ezh2 KO mice, we observed relative changes in H3K27me3 at specific loci, with some showing decreased H3K27me3 levels while other genes show increased levels. It will be interesting to explore specific genomic loci being more resistant or susceptible to epigenetic modifications holds true at critical fate-determining genes in other tissues, both in terms of normal development and with respect to neurodevelopmental and psychiatric diseases.
). Ezh2 is the primary methyltransferase targeting trimethylation of histone 3 lysine 27 (H3K27me3), which is critical for gene repression. In humans, EZH2 variants lead to Weaver Syndrome, a complex disease with varying degrees of intellectual disabilities. Loss of Ezh2 in mice can lead to premature neuronal differentiation, migration defects and changes in neuronal fate, but the role of Ezh2 in forebrain interneurons has not been explored. We demonstrated that loss of Ezh2 in the MGE reduced the overall number of MGE-derived interneurons in the cortex and caused a shift in fate with an increase in SST+ and a decrease in PV+ interneurons. Consistent with this result, single cell sequencing of the MGE revealed increased expression of SST-related genes cells in the KO MGE, whereas genes predictive of PV-fated interneurons were decreased. We found no changes in the intrinsic e-phys properties of PV+ or SST+ cells in the KO cortex. However, PV+ cells in the KO displayed more complex axonal arbors, with increased axonal length and arborization compared to WT PV+ cells. Despite a global reduction of H3K27me3 in Ezh2 KO mice, we observed relative changes in H3K27me3 at specific loci, with some showing decreased H3K27me3 levels while other genes show increased levels. It will be interesting to explore specific genomic loci being more resistant or susceptible to epigenetic modifications holds true at critical fate-determining genes in other tissues, both in terms of normal development and with respect to neurodevelopmental and psychiatric diseases.
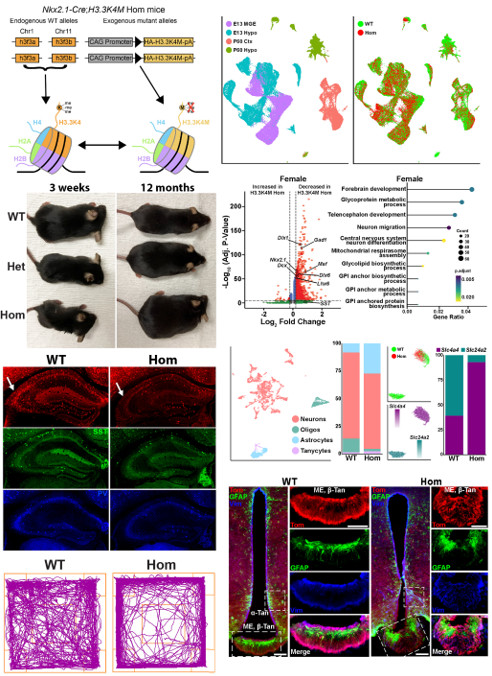
While H3K27me3 is critical for gene repression, methylation at histone 3 lysine 4 (H3K4me) is necessary for gene activation. Humans with variants in several methyltransferases that target H3K4me display a complex set of symptoms, but there is a comorbidity of epilepsy, intellectual disability, and short stature in these H3K4me-associated diseases. This phenotype is indicative of altered excitatory-inhibitory balance and hypothalamic defects in these diseases. We developed a creative genetic strategy to specifically reduce H3K4me levels in the MGE and hypothalamus during embryogenesis. These mutant mice have fewer forebrain interneurons, deficient network rhythmogenesis, and increased spontaneous seizures and seizure susceptibility. Mutant mice are significantly smaller than control littermates, but they eventually became obese due to striking changes in the genetic and cellular hypothalamus environment in these mice. Perturbation of H3K4 methylation in these cells produces deficits in numerous NDD-associated behaviors, with a bias for more severe phenotypes in female mice. Single cell sequencing reveals transcriptional changes in the embryonic and adult brain that underlie many of these phenotypes. In sum, our findings highlight the critical role of H3K4 methylation in regulating survival and cell-specific gene regulatory mechanisms in forebrain GABAergic and hypothalamic cells during neurodevelopment to control network excitability and body size homoeostasis. A preprint of this manuscript has been posted (Li…Petros bioRxiv 2025 ![]() ) and the manuscript is currently in revision.
) and the manuscript is currently in revision.
Mechanisms regulating fate and maturation of CGE-derived interneurons
Research over the last 20+ years has generated many insights in MGE development. However, our knowledge of mechanisms regulating CGE-derived interneurons lags significantly behind. While CGE-derived interneurons make up ~30% of cortical interneurons in the mouse, this population is significantly increased in humans and non-human primates, with the largest increase in VIP+ disinhibitory bipolar interneurons. There is evidence that CGE-derived interneurons may be a critical nexus for multiple neuropsychiatric disorders, and the CGE is the primary source of tumors and lesions in tuberous sclerosis complex (TSC). Thus, a better understanding of mechanisms regulating fate and maturation of CGE-derived interneurons is needed to understand both normal brain development and potential disease etiologies. Part of the lab is focused on this question.
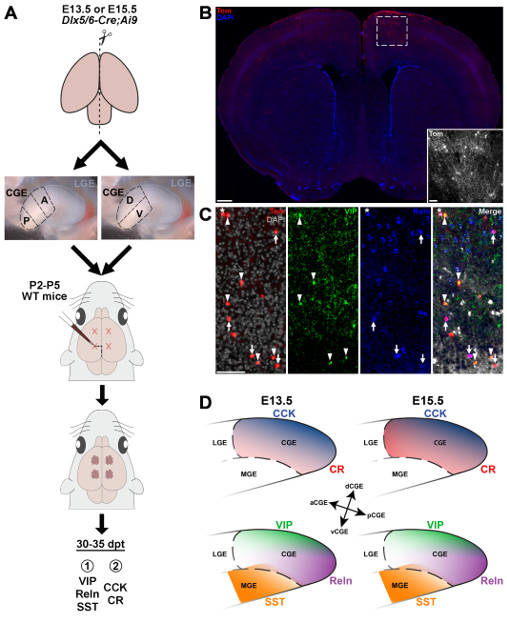
Transplantation studies focused specifically on the MGE revealed both a spatial and temporal logic relating MGE subdomains with mature interneuron fate: SST+ interneurons arise primarily from the dorsal-caudal MGE at earlier embryonic ages (E12.5-E15.5) whereas PV+ interneurons have a complimentary bias from the ventral-rostral MGE and are born throughout neurogenesis (E12.5-E18.5). A similar organization has never been explored in the CGE. To explore this relationship between origin and fate of CGE-derived interneurons, we harvested anterior, posterior, ventral and dorsal CGE cells from E13.5 and E15.5 Dlx6aCre;Ai9 mice and transplanted these cells into the cortex of P2-5 WT pups. Brains were harvested at P30-35 and immunostained for CGE-specific interneuron markers to correlate spatial and temporal origin with mature CGE-derived interneuron subtypes. Our results indicate that there are significant spatial biases in the CGE, with specific interneuron subtypes preferentially arising from distinct CGE subdomains: there is a strong bias for the dorsal CGE to generate VIP+ and CCK+ cells whereas Reln+ cells are biased to arise from the posterior CGE. These biases are relatively stable over time, with only CR+ interneurons showing increased production at E15.5, implying a minimal relationship between temporal birthdate and interneuron subtype. We are now exploring if any differentially expressed genes in these regions are critical for promoting these subtypes of CGE-derived interneurons. A preprint of this manuscript has been posted (Garg…Petros bioRxiv 2025 ![]() ) and submitted for publication.
) and submitted for publication.
The recent advent CRISPR/Cas based ‘Perturb-seq’ screens are a promising tool to identify genes involved in cell fate specification. We have developed an unbiased Perturb-seq approach using a Lentivirus gRNA library in combination with Cas9-expressing mESC-derived neural progenitors to identify TFs that promote CGE fates. These mESCs have been retrofitted with a fluorescent CGE reporter allowing us to identify CGE-fated cells. This experiment should identify candidate genes whose activation or suppression promote CGE fate for future investigation. This study is ongoing.
 BACK TO TOP
BACK TO TOP