Research
The central nervous system underlies all our experiences, actions, emotions, knowledge, and memories. With billions of neurons each firing hundreds of times per second, the complexity of the brain is stunning. To pare down the task of understanding something so complex, our research approach calls for studying the workings of a single central neuron—the pyramidal neuron from the CA1 region of the hippocampus.
Figure 1. Acute Hippocampal Brain Slice
(Click image to enlarge.)
The hippocampus is essential for long-term memory in humans and is among the first brain regions affected by epilepsy and Alzheimer’s disease. To understand how the hippocampus stores and processes information, we focus on one of its principal cell types, the CA1 pyramidal neuron. Each pyramidal neuron in the CA1 region of the hippocampus receives tens of thousands of inputs onto its dendrites, and it is commonly thought that information is stored by altering the strength of individual synapses (synaptic plasticity). Recent evidence suggests that the regulation of synaptic surface expression of glutamate receptors can, in part, determine synaptic strength. However, the dendrites contain an abundance of ion channels that are involved in receiving, transforming, and relaying information in the dendrites, adding an additional layer of complexity to neuronal information processing.
We found that the A-type potassium channel subunit Kv4.2 is highly expressed in the dendritic regions of CA1 neurons in the hippocampus and, as one of the primary regulators of dendritic excitability, plays a pivotal role in information processing. Kv4.2 is targeted for modulation during the types of plasticity thought to underlie learning and memory. Moreover, we found that the functional expression level of Kv4.2 regulates the subtype expression of NMDA–type glutamate receptors, the predominant molecular devices controlling synaptic plasticity and memory. We are currently following up on these findings with more detailed investigations into the mechanisms of activity-dependent Kv4.2 regulation and its role in neuronal development. In addition, we are investigating the role of dendritic voltage-gated channels in CNS disorders, including autism-spectrum disorder and Alzheimer’s disease.
Role of voltage-gated ion channels in synaptic development and disease
Kv4.2 control of firing patterns in hippocampal CA1 pyramidal neurons
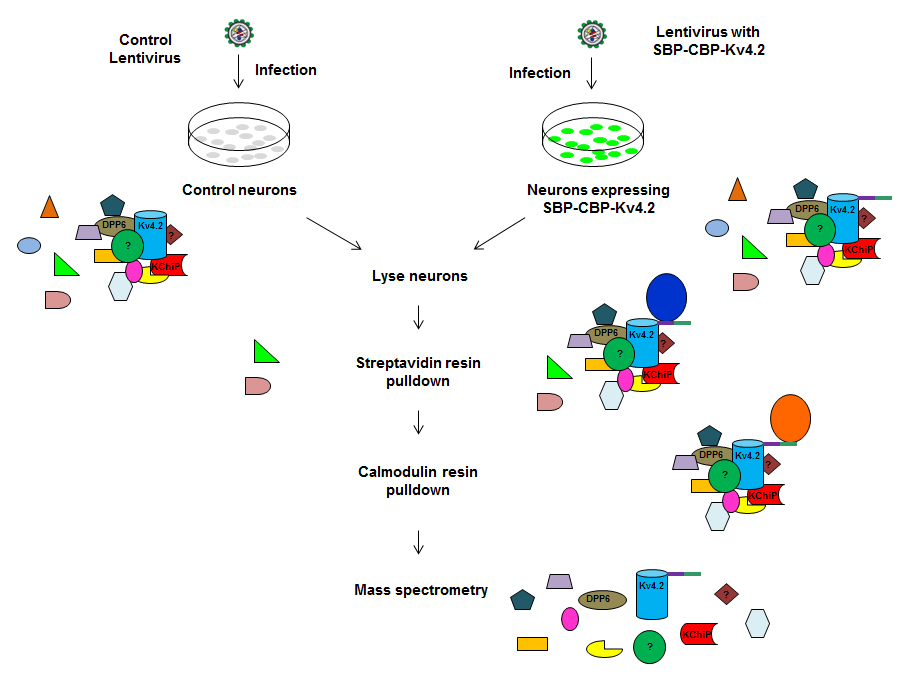
Figure 2. Schematic for Tandem Affinity Purification of Kv4.2 binding partners
(Click image to enlarge.)
Although recent molecular cloning studies found that several families of voltage-gated K+ channel genes are expressed in the mammalian brain, information about the relationship between the protein products of these genes and their various neuronal functions is still lacking. Our lab used a combination of molecular, electrophysiological, and imaging techniques to show that Kv4.2, an A-type voltage-gated potassium channel subunit, controls action potential (AP) half-width, frequency-dependent AP broadening, and dendritic AP propagation. More recently, we examined the role of A-type K+ channels in regulating synaptic plasticity, neuronal development and disease (1).
Neuronal excitability in Alzheimer's disease
Alzheimer's disease (AD), the most common form of dementia, is characterized by progressive neuronal loss, which eventually leads to death. Despite massive efforts over the last few decades, the etiology of AD is not well understood. A major challenge for AD research, and for the development of treatments, is that most AD patients are not diagnosed until neuronal function is irreversibly compromised. Therefore, it is crucial to identify neuronal changes at pre-clinical stages, which could provide a basis for early diagnosis and help identify novel therapeutic targets. Neuronal hyperexcitability occurs early in the pathogenesis of AD and contributes to network dysfunction in AD patients. Although the beta-amyloid (Aβ) hypothesis suggests that AD is caused by extracellular accumulation of insoluble Aβ plaques, mounting evidence suggests that synaptic and memory impairments are mediated by soluble Aβ. In collaboration with the Roberson lab at the University of Alabama, former postbaccalaureate fellow Ben Throesch, tested the hypothesis that Aβ-induced hyperexcitability originates in the dendrites. We found that dendrites, but not somata, of hippocampal neurons were hyperexcitable in mice adult mice overexpressing Aβ. The dendritic hyperexcitability was associated with selective depletion of Kv4.2, a dendritic potassium channel important for the regulation of dendritic excitability and synaptic plasticity.
In a separate project Jakob Gutzmann and former postdoctoral fellow Eun Young Kim investigated synaptic changes in young, 2-month-old transgenic mice that overexpress human amyloid precursor protein (hAPP). At this age, the mice do not exhibit Aβ plaque accumulation but show higher soluble Aβ levels than non-transgenic (NTG) mice. Our findings suggest NMDARs as a possible target for prevention or treatment for memory loss in early stages of AD. We are currently testing the effect of NMDAR subunit antagonists on the progression of AD at both the cellular and behavioral level.
The role of DPP6 domain in its localization and function
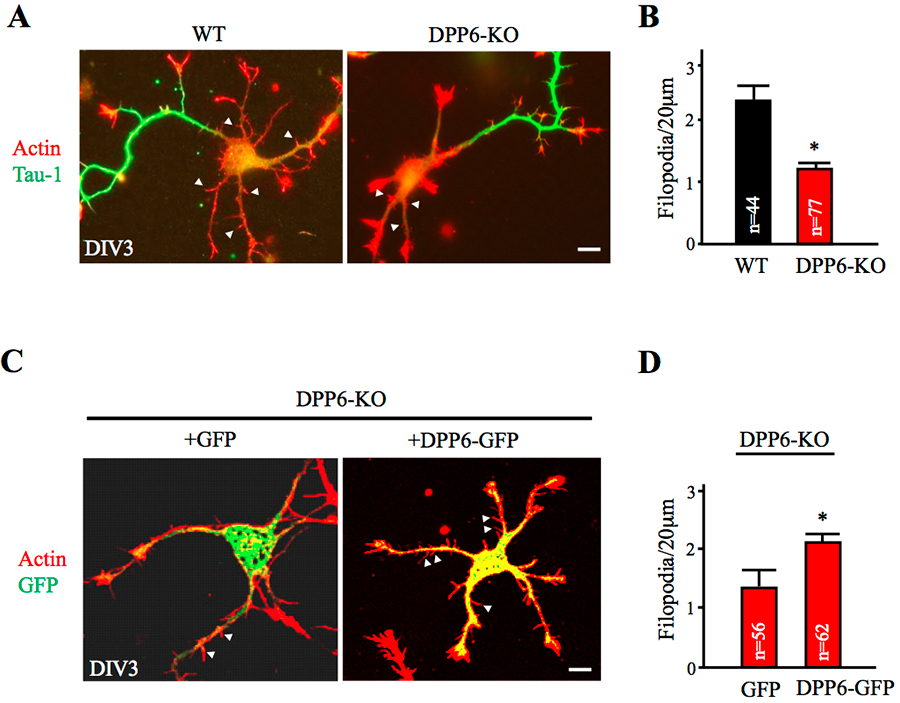
Figure 3. Dendritic filopodia formation is reduced in DPP6-KO hippocampal neurons and rescued by DPP6 expression.
(Click image to enlarge.)
(A,B) DIV3 wild-type (WT) or DPP6-knockout (KO) hippocampal neurons visualized for actin filaments with TRITC-phalloidin (1:1000, Sigma) and for axons with marker Tau-1 (1:1000, Millipore). The density of immature dendritic filopodia (arrowheads) is lower in the DPP6-KO neurons (n=77) than in the WT (n=44). (C,D) DIV3 DPP6-KO hippocampal neurons expressing either GFP control or DPP6-GFP, cultured for three days, and visualized for actin filaments with TRITC-phalloidin. The density of filopodia is higher in the DPP6-KO neurons that express DPP6-GFP (n=62) than in the GFP controls (n=56). Scale bars, 10μm. Error bars represent the mean±SEM; *p< 0.05.
Dipeptidyl peptidase-like protein 6 (DPP6) is an auxiliary subunit of the Kv4 family of voltage-gated K+ channels known to enhance channel surface expression and potently accelerate their kinetics. DPP6 is a single-transmembrane protein, which is structurally remarkable for its large extracellular domain. Included in this domain is a cysteine-rich motif, the function of which is unknown. Lin Lin found that the cysteine-rich domain of DPP6 is required for the protein's export from the ER and expression on the cell surface. Disulfide bridges formed at C349/C356 and C465/C468 of the cysteine-rich domain are necessary for the enhancement of Kv4.2 channel surface expression but not for DPP6's interaction with Kv4.2 subunits. The short intracellular N-terminal and transmembrane domains of DPP6 associate with and accelerate the recovery from inactivation of Kv4.2, but the entire extracellular domain is necessary to enhance Kv4.2 surface expression and stabilization. Our findings show that the cysteine-rich domain of DPP6 plays an important role in protein folding of DPP6, which is required for transport of DPP6/Kv4.2 complexes out of the ER.
We showed recently (1) that DPP6 regulates the formation and stability of dendritic filopodia during early neuronal development, which is independent of Kv4.2. In order to identify additional DPP6 binding proteins, a TAP purification approach was employed by Jiahhua Hu to isolate the DPP6 protein complex from hippocampal neurons. Mass-spectrometry analysis identified known proteins such as Kv4 family members and numerous novel synaptic proteins, which Jiahua Hu and Jung Park are currently examining.
Dendritic trafficking of voltage-gated calcium channels
We are currently investigating the expression and trafficking of the voltage-gated calcium channel Cav2.3. Cav2.3 is highly expressed in the dendrites of hippocampal and cortical neurons, where it is capable of generating large calcium spikes in response to both back-propagating action potentials and synaptic activity. Thus, alterations in Cav2.3 mRNA localization and translation could have a dramatic impact on cellular excitability and calcium signaling. Recent evidence suggests that Cav2.3 mRNA can be targeted by the Fragile-X mental retardation protein (FMRP), an mRNA–binding protein that regulates translation in dendritic spines. Loss of FMRP results in Fragile X Syndrome, the most common form of inherited intellectual disability in humans. We are therefore investigating the possibility that FMRP can regulate translation of Cav2.3 and will determine whether this regulation underlies aspects of Fragile X Syndrome.

Figure 4. Tissue under microscope with green laser light.
(Click image to enlarge.)
Toward this goal, Ying Liu performed real time PCR on mRNA isolated from the hippocampi or cortex of wild-type and FMRP knockout (KO) male mice and examined the mRNA levels of several dendritic proteins. When comparing hippocampi from FMRP–KO and wild-type mice at 3- or 8-weeks of age, she found no significant difference in the mRNA levels of Cav2.3, Kv4.2, PSD-95, and HCN-1. She will further characterize this regulation by identifying FMRP–binding sites on Cav2.3.
In conjunction with these experiments, Ivan Trang is determining how FMRP affects Cav2.3 protein expression. From synaptoneurosomes isolated from mouse cortex, he found that, at 3-weeks of age, Cav2.3 protein levels are lower in FMRP-KO mice than in wild-type mice. In addition, primary neurons cultured from FMRP–KO mice exhibit lower surface Cav2.3 levels than do wild-type mice. Thus, loss of FMRP leads to a reduction in both synaptic and surface Cav2.3 protein. To determine how this might affect neuronal physiology, Erin Gray will record Cav2.3–mediated calcium currents as well as basic firing properties from wild-type and FMRP–KO neurons.
While FMRP has a clear role in regulating mRNA stability, recent evidence suggests that FMRP may directly regulate the internalization and degradation of voltage-gated calcium channels. Given that little is known about the pathways that underlie Cav2.3 degradation, Erin Gray and former postbaccalaureate fellow Joshua Lee began experiments aimed at a better understanding of this process. In heterologous cells overexpressing Cav2.3, they showed that Cav2.3 undergoes activity-dependent ubiquitination and degradation by the proteasome. Gray has begun to further investigate a possible role for ubiquitin-mediated alterations in surface levels of Cav2.3 and plans to perform a variety of electrophysiological recordings to determine the physiological consequences of this regulation.
Co-regulation of HCN1 and Kv4.2
In CA1 pyramidal neuron dendrites, HCN channels, responsible for Ih current, and Kv4.2 channels, responsible for IA current, are critically important in signal processing and dendritic integration of synaptic inputs. Both channels show a similar pattern of distribution with an increased density from the soma to the apical dendrite. Using hippocampal primary cultured neurons, Emilie Campanac studied the potential co-regulation of HCN1 and Kv4.2. Results so far point to a reciprocal regulation, with overexpression of Kv4.2 being associated with an increase in Ih current density without any change in sodium and calcium current, while overexpression of HCN1 leads to an increased in IA current density. Pharmacological blockade of Ih with cesium (2mM) caused a reduction in both current densities. Our data strongly suggest homeostatic regulation between IA and Ihcurrents. We are currently investigating the molecular mechanism underlying this co-regulation.
 BACK TO TOP
BACK TO TOP