Research
…Continued from the Home Page:
Studies on translational control
Early accomplishments of the SNCGE in this area include discovering the novel regulatory mechanism that induces translation of GCN4 mRNA via small upstream ORFs (uORFs) in the mRNA leader by phosphorylation and inhibition of eIF2 by the kinase Gcn2, now understood to regulate expression of key transcription factors (Atf4 and Atf5) in mammals and implicated in learning and memory. We also made key contributions to identifying the eIF2α kinase Gcn2 and elucidating its regulation by amino acids, via allosteric control of kinase activity by uncharged tRNAs and Gcn2 regulatory domains, and by the TOR pathway through Gcn2-Ser577 phosphorylation. Our group discovered the Gcn2 positive effectors Gcn1 and Gcn20 and the importance of ribosomal Gcn1/Gcn20/Gcn2 regulatory complexes in Gcn2 activation. We identified the eIF2α phosphatases in yeast and made key contributions to elucidating the mechanism whereby phosphorylated eIF2 inhibits its GDP-GTP exchange factor, eIF2B, defining the catalytic and regulatory subcomplexes of eIF2B and their distinct roles in binding phosphorylated or nonphosphorylated eIF2. Our group contributed to understanding fundamental mechanisms of eukaryotic translation initiation by (i) determining the subunit composition and identifying in vivo functions of the yeast eIF3 complex and uncovering its multiple contacts with the 40S ribosome; (ii) identifying a higher-order assembly of initiation factors 1, 2, 3, and 5—the multifactor complex (MFC)—and elucidating its roles in PIC assembly and accurate start codon selection. We uncovered the functions of ABCE proteins Rli1/ABCE1 and Arb1 in PIC assembly and ribosome biogenesis, and identified the tRNA methyltransferase Gcd10/Gcd14, which contributed to the discovery of the TRAMP-mediated RNA surveillance pathway.
As noted above, a key accomplishment of the SNCGE was elucidating the unique mechanism of translational control of GCN4 mRNA. According to the current model, scanning ribosomes translate the 5′-most uORF (uORF1) and, under non-starvation conditions, reinitiate translation at downstream uORFs 2, 3, or 4 and subsequently dissociate from the mRNA, keeping GCN4 translation repressed. In starvation conditions, the reinitiating ribosomes bypass uORFs 2–4 and reinitiate at GCN4 instead, owing to lowered availability of the ternary complex (TC)—comprised of initiation factor 2 (eIF2), GTP, and initiator Met-tRNAi—which binds to the small (40S) ribosomal subunit to assemble a 43S preinitiation complex (PIC). TC abundance is reduced in starved cells by phosphorylation of eIF2α by Gcn2, converting eIF2 from substrate to inhibitor of its guanine nucleotide exchange factor (GEF) eIF2B. Hence, GCN4 translation is an in vivo indicator of impaired TC loading on 40S subunits. We previously exploited this fact to isolate mutations in subunits of eIF2B that constitutively derepress GCN4 (Gcd− phenotype) by lowering TC assembly in the absence of eIF2 phosphorylation. Subsequently, we used the Gcd− selection to identify domains/residues in eIF2 and tRNAi, eIF1, eIF1A, eIF3, and residues of 18S rRNA located near the 'P' decoding site of the 40S subunit, that participate in rapid TC recruitment in vivo (Figure 1A). In collaboration with Jon Lorsch's group, we demonstrated that segments/residues in eIF1, eIF1A, tRNAi and 18S rRNA, which are implicated genetically in TC recruitment, also stimulate the rate of this reaction in a fully reconstituted yeast in vitro translation system. These findings have helped to illuminate the molecular mechanism of TC recruitment and assembly of 43S PICS at an early stage of the initiation pathway.
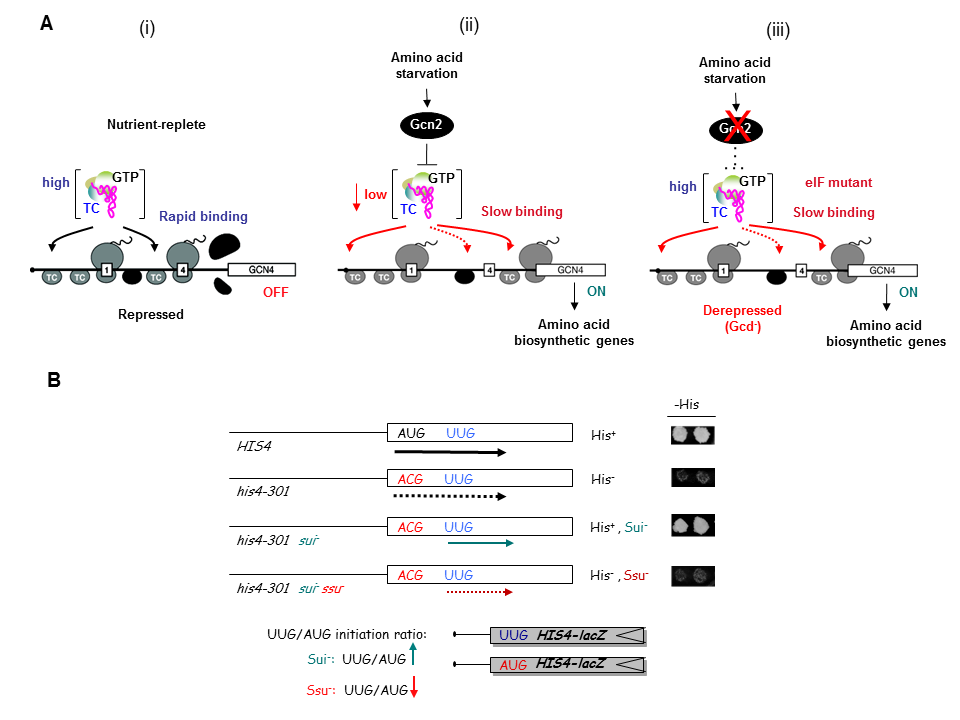
Click image to enlarge.
A network of molecular interactions in the preinitiation complex controls the fidelity of start codon selection during ribosomal scanning
We have also investigated the roles of various eIFs, tRNAi and the 40S subunit in scanning the mRNA 5′ untranslated region and in accurately identifying the AUG initiation codon. These studies exploit a genetic selection for mutations that elevate initiation at near-cognate UUG start codons (Sui− phenotype) or suppress this aberrant initiation event (Ssu− phenotype) (Figure 1B). By this approach, we showed that eIF1A and the c-subunit of eIF3 make critical contributions to accurate AUG recognition, and localized these functions to the unstructured N- and C-tails of eIF1A and the N-terminal domain of eIF3c. In collaboration with Lorsch's group, we provided strong biochemical evidence that dissociation of eIF1 from the scanning PIC is a critical step in AUG recognition, and that eIF1 serves as a 'gate-keeper' to suppress initiation at non-AUG triplets. Biochemical analysis revealed that eIF1 stabilizes an 'open' conformation of the 40S subunit conducive to scanning and loading of TC in a relatively unstable state (POUT), which enables inspection of successive mRNA triplets in the P site for complementarity with the anticodon of Met-tRNAi. However, eIF1 must dissociate from the PIC for AUG selection, as the absence of eIF1 favors the closed, scanning-arrested conformation of the 40S subunit with TC more tightly bound (PIN state). We established that basic residues in eIF1's helix-1 and β-hairpin-1 mediate 40S binding and stabilize the open, POUT complex to promote Met-tRNAi recruitment and accurate AUG selection in vivo. We also showed that 10–amino acid repeats in the C-terminal tail (CTT) of eIF1A (dubbed "scanning enhancers" or SE elements) function together with eIF1 to stabilize the open, POUT complex but must be antagonized by scanning-inhibitory (SI) elements in the N-terminal tail (NTT) and helical domain of eIF1A to achieve rearrangement to the closed, PIN complex for start codon recognition. In collaboration with Lorsch's group, we established that the eIF1A CTT moves towards the N-terminal domain (NTD) of eIF5 in response to AUG recognition, and that this movement—which depends on the eIF1A SE elements—is required for completion of GTP hydrolysis in the TC and release of Pi from eIF2-GDP·Pi. We proposed that accommodation of tRNAi in the PIN state triggers eIF1 displacement, which in turn triggers movement of the eIF1A-CTT and Pi release (Figure 2). We also obtained physical evidence for conformational rearrangements in the PIC on AUG recognition, from altered patterns of directed hydroxyl radical cleavage (DHRC) of rRNA by Fe(II)-BABE tethered to cysteine residues in eIF1A between PICs reconstituted with AUC or AUG start codons, indicating constriction of the mRNA entry channel, tighter Met-tRNAi binding in the PIN state, dissociation of eIF1 from the 40S, and displacement of the eIF1A-CTT from the P-site on AUG recognition.
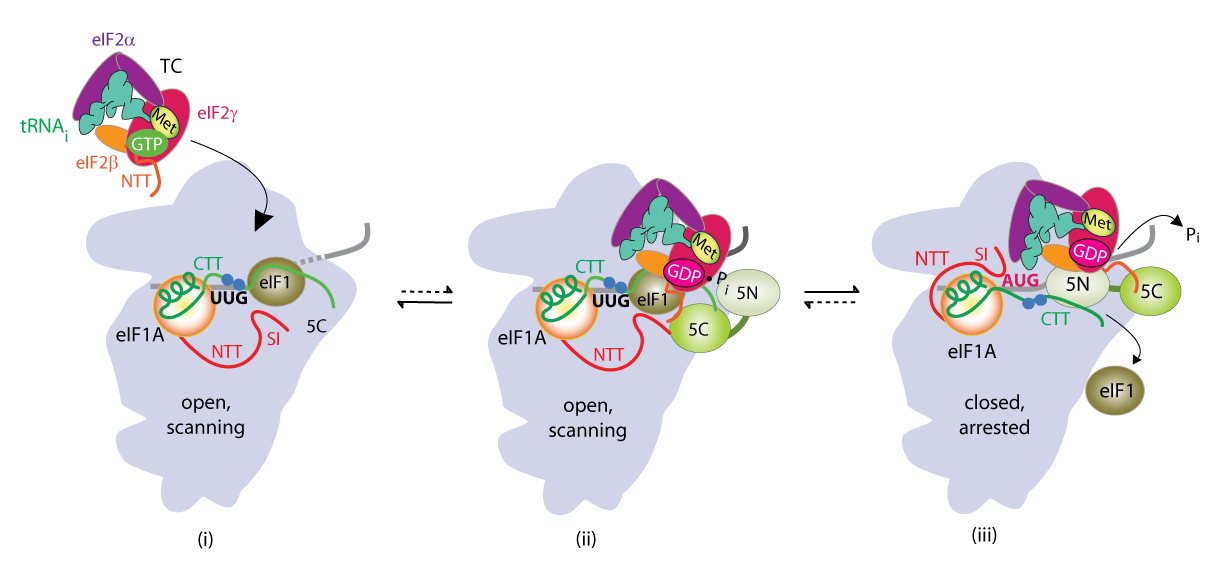
Click image to enlarge.
Collaboration with Venki Ramakrishnan’s group and the Lorsch lab has yielded cryo-EM reconstructions of yeast PICs representing different stages of the initiation pathway, including an open complex (py48S-open) formed using mRNA with AUC start codon, and a closed complex (py48-closed) with an AUG codon. The py48S-open PIC displays a widened mRNA entry channel conducive to mRNA binding and scanning, and an incompletely formed P site lacking 40S-tRNAi contacts, consistent with a POUT state. Downward movement of the 40S head in the closed complex clamps the mRNA into the binding cleft and encases the tRNAi in a fully formed P site, consistent with a PIN state. Comparing the open and closed PIC structures revealed numerous molecular interactions occurring exclusively in only one of the two states, suggesting that these interactions preferentially favor only one of the two states. We have confirmed these predictions by showing that mutations in the relevant factors or 40S proteins that perturb the interactions confer phenotypes diagnostic of specific destabilization of the open complex (Sui- and Gcd- phenotypes) or the closed complex (suppression of Sui-, or Ssu- phenotypes). Analyzing these mutants in the yeast reconstituted system confirmed that they alter the rate or stability of TC binding to PICs reconstituted with mRNAs harboring AUG or UUG start codons in the manner predicted from their in vivo phenotypes. In this way, we demonstrated a network of interactions revealed in the PIC structures that specifically favor the open complex, including interactions of 40S exit channel protein uS7 with eIF2α, eIF1 contacts with eIF2β, eIF2β contacts with the anticodon loop of tRNAi, and predicted clashes of eIF1 and eIF2β with the tRNAi D-loop in its location in the closed complex. Similarly, we showed that other interactions specifically favor the closed state, including interactions of the eIF1A-NTT with tRNAi, of 40S entry channel protein uS3 with mRNA, and distinct uS7-eIF2α contacts (Figure 3).
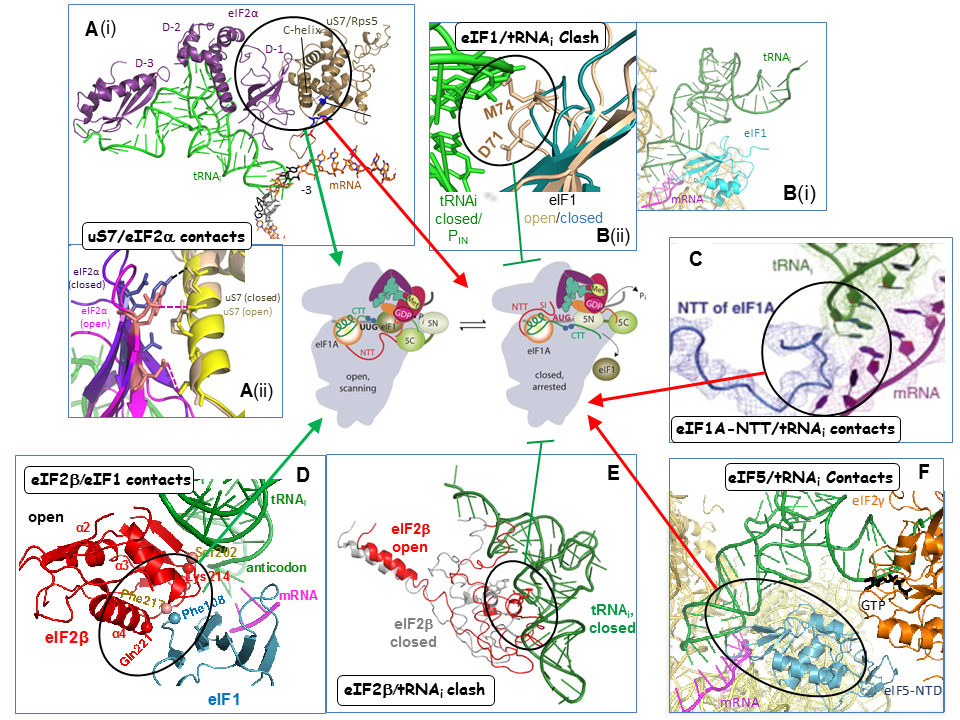
Click image to enlarge.
A more recent structure from the Ramakrishnan group revealed a new closed complex with the N-terminal domain (NTD) of eIF5 bound to the 40S subunit in place of eIF1. Rather than clashing with Met-tRNAi, as predicted for eIF1, the eIF5-NTD makes favorable interactions that stabilize a more fully accommodated PIN state. In collaboration with the Lorsch and Adesh Saini labs, we showed that eIF5 substitutions that should stabilize or destabilize the eIF5-NTD/tRNAi interface have the predicted respective Sui- or Ssu- phenotypes, and also the predicted opposite effects on the rate of Pi release from eIF2·GDP·Pi in reconstituted PICs. Thus, the eIF5-NTD stabilizes Met-tRNAi binding in the P site and blocks re-binding of eIF1, enabling Pi release and preventing the resumption of scanning at AUG codons.
Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A, and functional cooperation between Ded1 and its paralog Dbp1.
In addition to the ribosome-binding initiation factors and 40S proteins that control conformational transitions within the PIC, an equally important aspect of ribosomal scanning concerns the roles of mRNA-binding initiation factors in stimulating PIC attachment to the mRNA 5’ end and subsequent scanning through structured 5’UTRs, including the cap-binding complex eIF4F, helicase eIF4A and co-factor eIF4B, poly(A)-binding protein (Pab1), and auxiliary RNA helicases Ded1 and Dbp1. In previous studies, in collaboration with the Lorsch group, We dissected the NTD of eIF4G—the scaffolding subunit of eIF4F that harbors binding sites for eIF4E, PABP, and eIF4A—and identified conserved elements (RNA1, boxes 1–3) that functionally overlap to promote eIF4G interaction with mRNA and PABP, thereby activating mRNA for PIC attachment. We also dissected the functional domains of eIF4B and identified a novel role for this factor in promoting eIF4A/eIF4G association; and we further demonstrated binding of eIF4B near the mRNA entry channel of the 40S subunit, which likely contributes to eIF4F function in PIC recruitment to the mRNA.
More recently, we have conducted ribosome footprint profiling of mutants defective or lacking in eIF4A, eIF4B, Ded1 or Dbp1 to identify and characterize native mRNAs hyperdependent on each factor for efficient translation in vivo. eIF4A and Ded1 both have been implicated in resolving mRNA structures that impede ribosome attachment or scanning to the start codon, but whether they perform distinct functions in vivo was poorly understood. Despite similar reductions in bulk translation by ded1 and tif1 (eIF4A) mutations, ribosome profiling showed that ded1 mutations reduce the translational efficiencies (TEs) of >600 mRNAs, whereas inactivation of eIF4A similarly affected <40 mRNAs. Ded1-dependent mRNAs show greater than average 5’UTR length and propensity for secondary structure, implicating Ded1 in PIC attachment or scanning though structured 5' UTRs on hundreds of mRNAs. eIF4A, by contrast, promotes a step of initiation common to nearly all mRNAs regardless of their 5’UTR structures. While eIF4B is generally regarded as an eIF4A cofactor, ribosome profiling showed that eliminating eIF4B preferentially impairs translation of mRNAs with long structured 5’UTRs, and reduces the relative TEs of many more genes than does inactivation of eIF4A, supporting an eIF4A-independent role for yeast eIF4B. We also found that eIF4B, eIF4A, and Ded1 mutations also preferentially impair translation of longer mRNAs in a manner that could be mitigated by their high-level association with eIF4F and Pab1, as revealed by published RIP-seq analysis of these proteins. As eIF4F and Pab1 stabilize the “closed-loop” mRNP generated by eIF4G-PABP interaction, efficient closed-loop formation can likely compensate for reduced functions of eIF4B, eIF4A, and Ded1 in 48S PIC assembly.
In collaboration with the Lorsch lab, we have reconstituted the acceleration of 48S PIC assembly on structured native mRNAs by Ded1, and also shown that this depends on Ded1 interactions with eIF4F. Measuring the kinetics of 48S PIC assembly in the yeast reconstituted system for native mRNAs identified as either hyper- or hypodependent on Ded1 in vivo, we found that while eIF4A is essential for all mRNAs, Ded1 hypodependent mRNAs can be recruited rapidly without Ded1, and Ded1 only moderately accelerates their recruitment. Ded1 hyperdependent mRNAs, by contrast, are recruited poorly without Ded1 and Ded1 greatly accelerates their recruitment. Stem-loop (SL) structures in the 5’UTRs were shown to confer Ded1-dependent recruitment on several hyperdependent native and synthetic mRNAs. We also found that eliminating domains in Ded1 that mediate its association with eIF4A or eIF4G increased the Ded1 concentration required for maximal rate acceleration and also decreased the maximal rate achieved with Ded1 for certain hyperdependent mRNAs. These results showed that Ded1 accelerates 48S assembly by resolving 5’UTR structures in a manner stimulated by interaction with eIF4F and eIF4A (Figure 4).
By ribosome profiling of mutants lacking the Ded1 paralog Dbp1, either alone or in combination with a ded1 mutation, we found that Dbp1 functionally cooperates with Ded1 throughout the translatome in stimulating translation of mRNAs with long, structure-prone 5’UTRs, and that the presence Dbp1 can mask the involvement of Ded1 on many such mRNAs. We also showed that Dbp1 mimics Ded1 in conferring greater acceleration of 48S PIC assembly in the yeast reconstituted system on mRNAs harboring structured 5’UTRs. Using the recently developed method for genome-wide profiling of 40S subunits, enabling quantification of 43S PIC within 5’UTRs, we provided direct in vivo evidence that Ded1 and Dbp1 stimulate scanning through structured 5’UTRs to enhance translation of Ded1- or Dbp-hyperdependent mRNAs, and we also uncovered cooperation between these helicases in promoting 43S PIC attachment to a subset of such mRNAs.
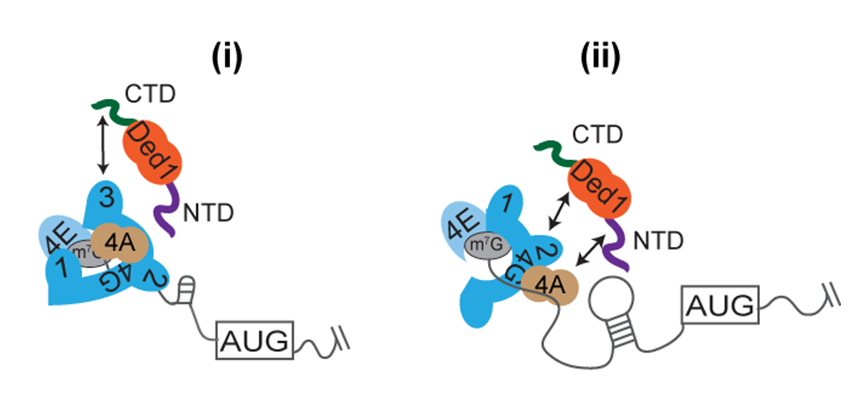
Click image to enlarge.
Conserved mRNA-granule component Scd6 targets Dhh1 to repress translation initiation and activates Dcp2-mediated mRNA decay in vivo
Scd6 protein family members are evolutionarily conserved components of mRNA granules. Scd6, and two other proteins containing RGG domains, Sbp1 and Npl3, were previously implicated as general translational repressors that function by binding to the RNA2 or RNA3 domains of eIF4G to inhibit 43S PIC recruitment by eIF4F·mRNPs. Scd6 also interacts with Dcp2, catalytic subunit of the decapping enzyme; and with the decapping activators Pat1 and the helicase Dhh1, both also implicated as general repressors of translation. To obtain evidence that Scd6, Sbp1, or Npl3 can act as translational repressors on particular mRNAs in vivo, we examined the effects of tethering MS2 fusions of each protein to a GFP reporter mRNA containing 3’UTR MS2 binding sites, either in WT cells or in mutants lacking DCP2, DHH1, or both genes. We found that tethering Scd6, but not Sbp1 or Npl3, repressed reporter mRNA abundance dependent on Dcp2 and also suppressed reporter mRNA translation via Dhh1. Repression of both GFP protein and mRNA was enhanced in a ccr4Δ mutant, lacking the major mRNA deadenylase activity of Ccr4-Not complex, suggesting that Ccr4 interferes with a more efficient repression pathway enlisted by tethered Scd6. Importantly, ribosome profiling and RNA-Seq analysis of scd6Δ and dhh1Δ mutants, and of the corresponding double mutants also lacking DCP2, suggests that Scd6 cooperates with Dhh1 in translational repression as well as turnover of a group of native mRNAs, and that both processes require DCP2, as the derepression of mRNA abundance and TEs conferred by deletion of SCD6 or DHH1 are suppressed by dcp2Δ. These last findings suggest that translational repression, in addition to accelerated mRNA degradation, conferred on native mRNAs by Scd6 and Dhh1 depend on mRNA decapping, leading to a pool of uncapped but relatively stable mRNAs with low TEs owing to a failure to recruit eIF4F.
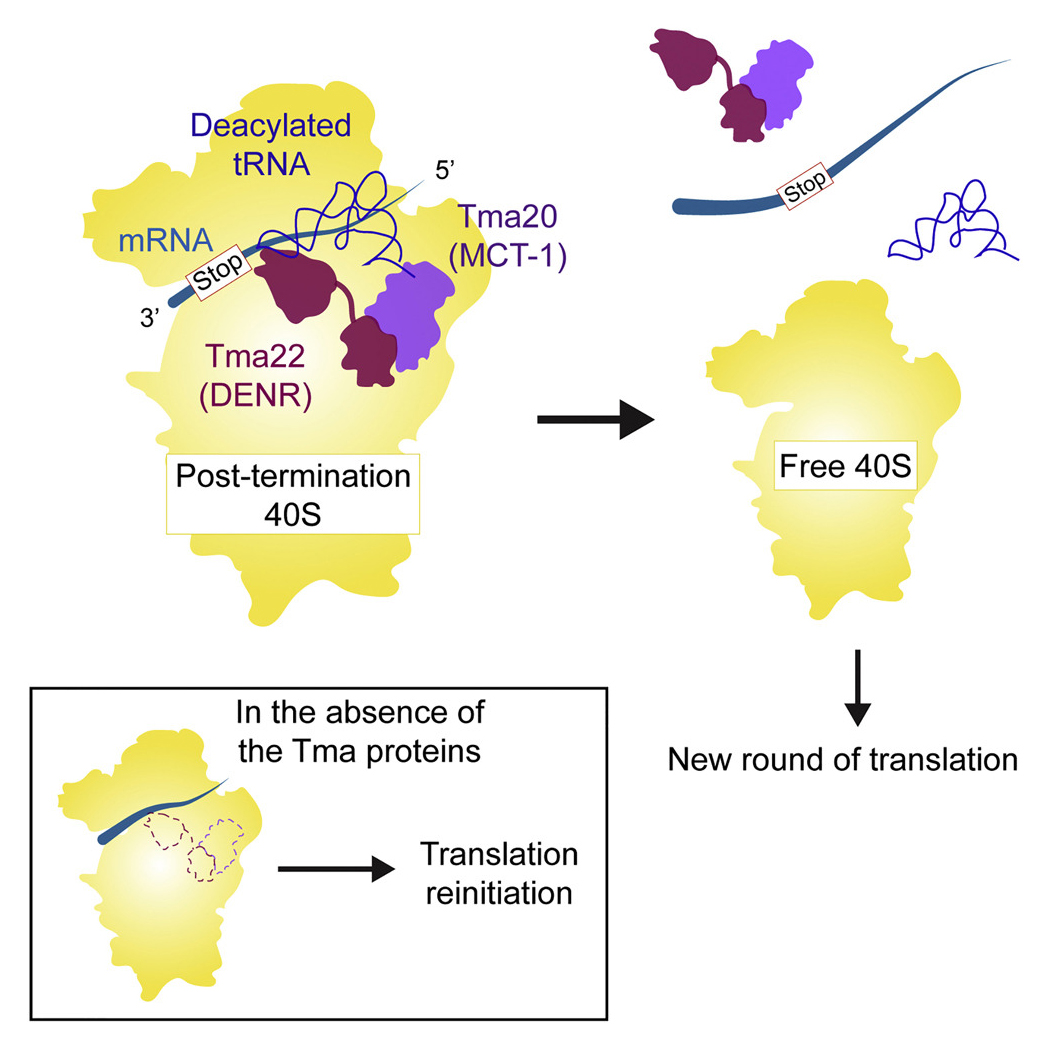
Rli1/ABCE1 and factors Tma64 (eIF2D), Tma20 (MCT-1), and Tma22 (DENR), catalyze successive steps in recycling of terminating ribosomes and suppress translation reinitiation in 3′ UTRs in vivo.
Ribosome recycling occurs after release of completed polypeptides at stop codons and involves dissociation of the 60S subunit from the 80S post-termination complex (post-TC). The protein Rli1/ABCE1 (ATP-binding cassette protein E1) catalyzes this reaction in vitro, but it was unknown whether Rli1 is essential for recycling in vivo. Ribosome profiling of an rli1 mutant depleted of Rli1, in collaboration with Rachel Green’s lab, revealed accumulation of 80S ribosomes at the stop codons and adjoining 3′ UTRs of most genes, consistent with defective recycling that allows ribosomes to proceed into 3′ UTRs. The presence of footprint peaks at 3′ UTR stop codons in all three frames, and the occurrence of footprint peaks at 3’UTR histidine codons on histidine starvation, on Rli1 depletion indicated that the 3’UTR ribosomes are translating. Predicted 3′ UTR translation products were detected on Rli1 depletion by Western analysis of epitope-tagged 3’UTR ORFs, whose small sizes and sequences determined by mass spectrometry indicated that un-recycled 80S post-TCs migrate a short distance from the stop codon and reinitiate translation without apparent codon preference. Analyzing a double mutant also lacking the ribosome-rescue factor Dom34 showed that Dom34 rescues the unrecycled 80S ribosomes generated on Rli1 depletion. These findings showed that Rli1 is required for ribosome recycling in vivo and acts globally with Dom34 to prevent aberrant reinitiation and translation of 3′ UTRs.
More recently, in collaboration with Nicholas Guydosh’s group, we provided evidence that yeast Tma64 (eIF2D), Tma20 (MCT-1), and Tma22 (DENR) function in the second step of ribosome recycling—the dissociation of mRNA and the tRNA that decoded the last sense codon from the 40S post-termination complex (40S post-TC). Previous biochemical studies showed that Tma64(eIF2D) or the related heterodimer Tma20 (MCT-1)/Tma22 (DENR), can catalyze this reaction in vitro, but evidence was lacking that they are important for ribosome recycling in vivo. By ribosome profiling double mutants lacking Tma64 and either Tma20 or Tma22, we observed 80S ribosomes queued immediately upstream of stop codons, consistent with a genome-wide block in 40S recycling with 40S post-TCs stalled at stop codons. We also obtained strong evidence that unrecycled 40S post-TCs in the mutants can reinitiate translation at AUG codons located downstream in 3’UTRs, as indicated by 80S peaks on such AUG codons in the profiling data; and by detecting epitope-tagged 3’UTR translation products whose expression was reduced on eliminating their presumptive 3’UTR AUG start codons. In vitro translation experiments done in collaboration with Sergey Dmitriev using reporter mRNAs containing upstream ORFs (uORFs) established that reinitiation at coding sequences downstream of the uORFs increased in mutant cell extracts devoid of the Tma factors, in a manner rescued by adding back the WT Tma proteins. These results support a crucial role for eIF2D/Tma64 or the Tma20(MCT-1)/Tma22(DENR) heterodimer in recycling 40S post-TCs at stop codons and thereby suppressing translation reinitiation in 3’UTRs (Figure 5).
Click image to enlarge.
Studies on transcriptional control
In our earlier work in the area of transcriptional control, we previously defined multiple clusters of hydrophobic residues that constitute the transcriptional activation domain of Gcn4, identified co-activators required for gene activation by Gcn4 in vivo, and defined a molecular program for recruitment of nucleosome modifying and remodeling enzymes and adaptor proteins that remove repressive chromatin structure and recruit TATA–binding protein and Pol II to Gcn4 target promoters. We also demonstrated co-transcriptional recruitment of the histone acetyltransferase (HAT) complexes SAGA and NuA4 to transcribed coding sequences and showed that they cooperate to enhance transcription-associated histone eviction and Pol II elongation in vivo. We described a two-stage recruitment mechanism for NuA4, involving the serine-5–phosphorylated C-terminal domain (CTD) of Pol II and methylated histones, and more recently we extended this mechanism to include the histone deacetylase (HDA) complex Rpd3C(S). In addition, we showed that the HDAs Rpd3, Hos2, and Hda1 have overlapping functions in deacetylating coding-sequence nucleosomes and obtained evidence that histone acetylation is a key determinant of co-transcriptional nucleosome disassembly. We further elucidated roles of the Pol II CTD kinases Kin28 and Bur1 and the C-terminal repeats (CTRs) of the Spt5 subunit of DSIF in recruitment of transcription elongation factor Paf1C. We showed that Ser5-CTD phosphorylation by Kin28 enhances recruitment of Bur1 near promoter regions where Bur1 contributes to Ser2–CTD phosphorylation. We then established that Kin28 enhances Paf1C recruitment by: (1) promoting Bur1 recruitment via the phosphorylated Pol II CTD, with attendant phosphorylation of Spt5 CTRs by Bur1; and (2) collaborating with Bur1 to generate Ser2–/Ser5–diphosphorylated Pol II CTD repeats. The resulting phosphorylated CTD and CTR repeats bind to distinct Paf1C subunits to enable efficient recruitment of the entire complex to elongating Pol II.
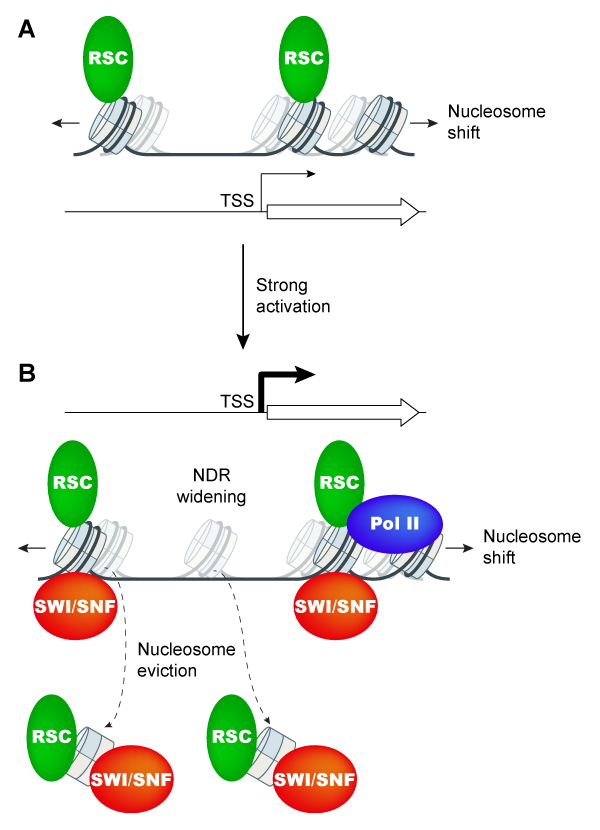
Functional interplay between chromatin remodelers SWI/SNF and RSC in evicting promoter nucleosomes at Gcn4-induced genes.
Most yeast genes transcribed by Pol II contain a nucleosome-depleted region (NDR) upstream of the CDS flanked by positioned “-1” and “+1” nucleosomes, with the transcription start site (TSS) located within the +1 nucleosome (+1_Nuc). Given its proximity to the TSS and upstream promoter elements, the +1_Nuc can interfere with formation of the PIC and initiation of transcription, and presumably must be evicted or repositioned downstream to facilitate transcriptional activation. A key unsolved aspect of transcriptional activation by Gcn4 is how it mediates the eviction of the nucleosomes that occlude promoter DNA sequences and block access by GTFs and Pol II. Indeed, the mechanism of this key step of gene activation and the impact of defective nucleosome eviction on transcription are not fully understood for any yeast activators. Previous studies implicated histone chaperones, chromatin remodelers (CRs), and HATs in the remodeling or eviction of nucleosomes from the promoters of certain yeast genes, but it was unclear whether these co-factors function broadly in nucleosome eviction, given that co-factor requirements at most yeast promoters are unknown. Eviction of promoter nucleosomes is considered to be rate-limiting for transcriptional activation, but the consequences of impaired nucleosome eviction on transcription have not been thoroughly analyzed genome-wide. We have been addressing these questions by analyzing histone H3 eviction for the hundreds of genes in the Gcn4 transcriptome on induction of Gcn4 in a panel of mutants lacking one or more co-factors implicated at particular yeast genes.
Using histone H3 chromatin immunoprecipitation of sonicated chromatin coupled to deep-sequencing (H3 ChIP-Seq) of single, double, and triple mutants lacking the catalytic subunit of the CR SWI/SNF (Snf2), HAT Gcn5, or general chaperone Ydj1, we previously uncovered collaboration among these co-factors in evicting the -1 and +1 nucleosomes and preventing nucleosome assembly in the intervening NDR at a large fraction of Gcn4 target genes. These co-factors also cooperate in H3 eviction at most other yeast promoters. The defects in promoter nucleosome eviction were associated with marked reductions in transcription only at the most highly expressed yeast genes, which includes the Gcn4 target genes when Gcn4 is induced, suggesting that promoter nucleosome eviction tends to be rate-limiting only at the most highly transcribed genes in yeast.
Having found that nucleosome eviction at Gcn4 target genes is only partially impaired in snf2Δ cells, we next asked whether the CR RSC participates with SWI/SNF in promoter nucleosome eviction. Previous studies showed that RSC acts broadly to maintain the correct positions of the -1_Nuc and +1_Nuc, and hence, proper NDR widths. ChIP-seq analysis of both H3 and Pol II on mutants lacking Snf2, depleted of the catalytic subunit of RSC, or impaired for both CRs, revealed additive defects in promoter nucleosome eviction of the 70 target genes exhibiting the most extensive nucleosome remodeling in WT cells on loss/depletion of SWI/SNF and RSC, with commensurate reductions in transcription. Conducting H3 ChIP-Seq of MNase-digested (versus sonicated) chromatin to achieve greater positional resolution, we uncovered a previously undetected widening of NDRs by >20bp at induced Gcn4 target genes in WT cells achieved through repositioning of both the -1 and +1 nucleosomes; and went on to establish that SWI/SNF and RSC cooperate in achieving wide, nucleosome-depleted NDRs at these induced genes. Cooperation between SWI/SNF and RSC in nucleosome eviction, positioning, and transcription was also observed at the small subset of ~200 most highly transcribed, constitutively expressed genes, suggesting a general cooperation by these CRs in achieving high-level transcription. ChIP-chip analysis indicated enrichment for both CRs at the most highly expressed and Gcn4-induced genes, supporting their direct roles at these genes. These results demonstrate extensive functional interplay between SWI/SNF and RSC in eviction and repositioning of promoter nucleosomes specifically at the most highly transcribed genes in yeast (Figure 6).
Click image to enlarge.
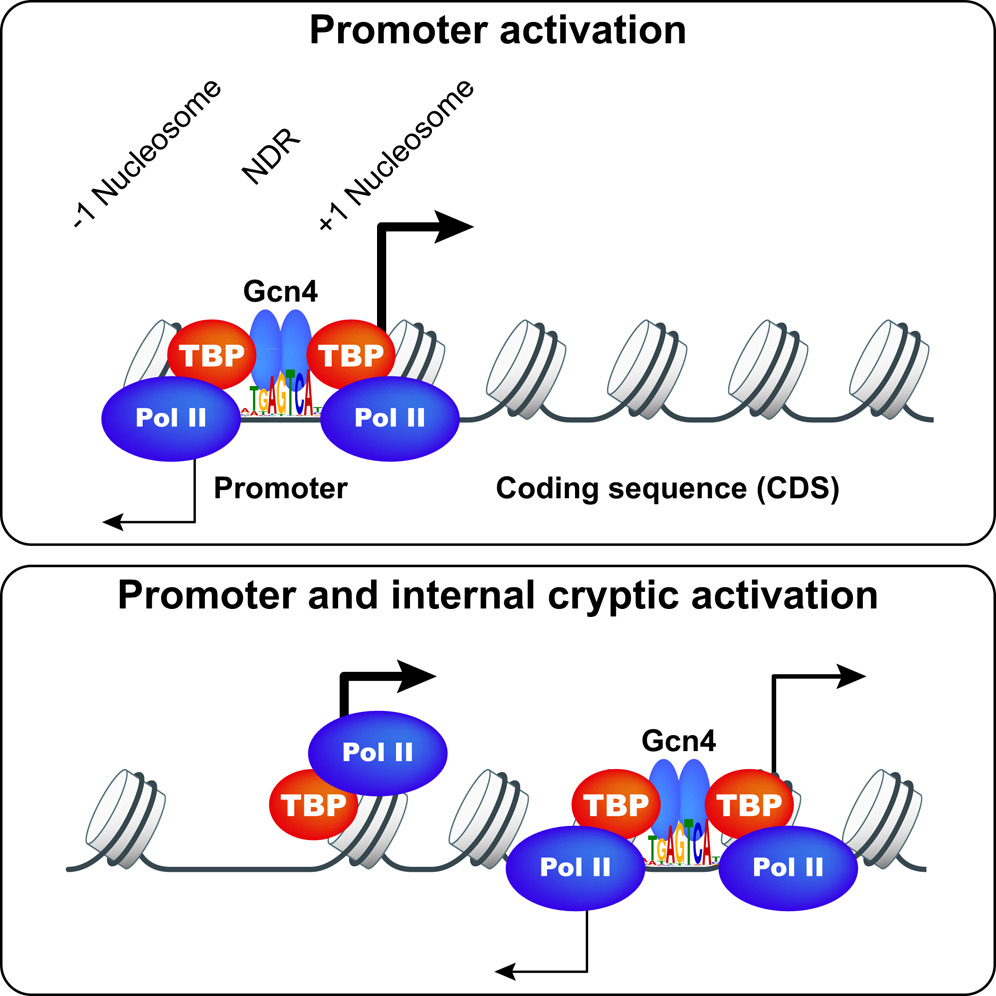
Gcn4 binding within coding regions can activate both internal and canonical 5’ promoters in yeast
Gcn4 and other yeast activators bind to upstream activation sequences (UASs), typically found within NDRs, but the genome-wide profile of Gcn4 binding was not unknown. Gcn4 ChIP-seq was conducted to identify all genomic Gcn4 binding sites in cells induced for Gcn4 synthesis by amino acid starvation. We identified 546 genomic sites occupied by Gcn4, with the peak densities centered on the known binding motif, which represents only ~30% of all significant matches to the Gcn4 binding motif in the genome. Analysis of nucleosome occupancy data from our previous MNase-seq analysis of starved cells revealed that the distance of a binding motif from the nearest nucleosome dyad, and its match to the consensus sequence, are the major determinants of Gcn4 binding in vivo. Surprisingly, only ~49% of genes showing Gcn4 binding contain the binding site in the conventional (C) location upstream of the TSSs, and analysis of both genome-wide mRNA expression data and our RNA Pol II ChIP-seq data in starved cells revealed that only ~70% of these conventional, Gcn4 occupancy peaks appear to activate transcription; thus indicating extensive negative control over Gcn4 function. Remarkably, ~300 genes exhibit Gcn4-binding within coding sequences (CDS), of which ~75 also exhibit Gcn4-induced transcription and contain only one such unconventional (UC) Gcn4 binding site in the vicinity. RNA-seq analysis revealed that many such unconventional (UC) Gcn4 peaks map between divergent antisense (AS) and sub-genic sense (SGS) transcripts originating from within the CDS that are induced by starvation. A subset of these UC Gcn4 peaks also reside just upstream of twin TBP occupancy peaks that we detected by TBP ChIP-seq, occurring only under inducing conditions. These findings established Gcn4 activation of cryptic, bidirectional internal promoters at the UC genes. Mutational analysis of ten UC genes confirmed that the Gcn4 binding sites (GBSs) within the CDS activate both the sub-genic and full-length transcripts from the same or adjacent genes. These results demonstrate that functional Gcn4 binding is not confined to UAS elements, and establish that cryptic, internal promoters can be activated by a well-defined TF.
Click image to enlarge.
 BACK TO TOP
BACK TO TOP