Virtual Quantitative Nanoscopy
In this section we will discus a new area of research the SAFB has just begun. In collaboration with a group led by Dr Balaban at the NHLBI, we began working on quantification of photonic behavior in Two-Photon Microscopy. This has led to other collaborations with Dr Boukari of the Section on Cell Biophysics in quantifying photon behavior in the case of Fluorescence Correlation Spectroscopy. It ties in well with another project we have ongoing in collaboration with Dr Ilev at the FDA. This latter work involves nanoscale imaging beyond the diffraction limit.
Quantification in Two Photon Microscopy
Recent developments in Two-Photon imaging have been aimed at pushing back the depth limit of this exciting imaging technique. Here at the SFAB we have been working in collaboration with a group at NHLBI led by Dr Balaban. This work has involved the development and quantification of a new approach to enhance Two-Photon microscopy. The basic tenet of the approach is to obtain Total Emission Detection (TED), as opposed to the usual epi/trans detection of two-Photon Imaging. An instrument called 'MoreLight' has been developed which uses optics to capture light from all directions as opposed to some small solid angle usually used in the technique.
To quantify this instrument the SFAB developed a simple emission-only fluorescence Monte-Carlo model. Here we are sealing with scattered light as we are handling light emitted through tissue so a photonic model is used. We can safely ignore the diffraction blurring caused as we are at the boundary of photonic/Wave behavior of light as we are only considering the Gain of the instrument. Initial results from our algorithm were shown to match well with data from a phantom study (given a scaling factor based on known instrumental losses due to efficiency of optical components), as shown in figure a. In figure b we show some initial calibration of the instrumental effectiveness in different tissue types at different depths.
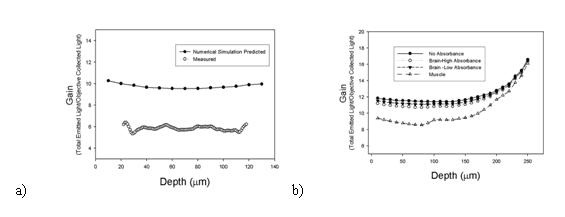
b) A graph showing how the model can be used to predict gains in different tissue types.
Correcting for Scattering in fluorescence Correlation Spectroscopy (FCS)
This collaboration has led to further work being done in collaboration with the section on Cell Biophysics. Here we aare helping to quantify the effects of scattering in FCS. In FCS it is common to use micro-beads to examine diffusion rates of a molecule, however at this time the effect this has on the imaging is not properly considered. It is generally noted that some blurring will occur, but no quantification of this is currently available. To examine this we have created a second Monte-Carlo simulation looking at the source fieled for a con-focal instrument. We have noted that the effects of scatter on an FCS illumination field are more complex than just blurring. The scattering (and relative anisotropy) have significant effects on the shape and location of the illuminated region. This implies effects on both where we are looking, but may also change the relative volumes under consideration, an effect which may bias the diffusion rate being monitored. In the figure below we show how scattering can completely destroy the confocal spot, and how relatively high anisotropy factors can reduce this effect. It becomes important then to understand the size and refractive index of the microbeads used and how these will effect the illumination field in FCS.
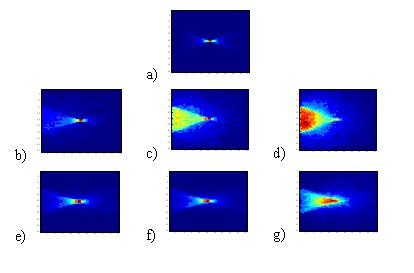
Nanoscopic Imaging Beyond the diffraction barrier
Among the modern biophotonics imaging techniques, high-resolution confocal laser microscopy is an intensely active field because this technique provides sharp, high-magnification, three-dimensional imaging with submicron resolution by noninvasive optical sectioning. However, in the subwavelength nanometric range, where there has recently been a great impetus for imaging and sensing intracellular structures and functions as well as for obtaining quantitative chemical information for light-tissue interactions at cellular/intracellular levels, the existing techniques have a major drawback related to the fundamental Rayleigh diffraction barrier.
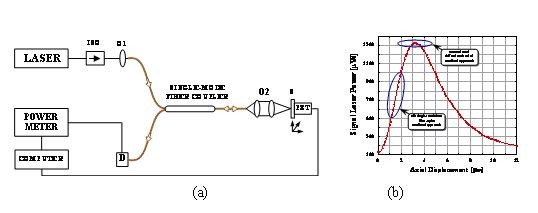
We have explored an effective way to avoid the resolution limit by employing a fiber-optic based confocal microscope approach which features a number of significant advantages in terms of ultrahigh-spatial resolution, diffraction-free laser delivery, flexibility, and scanning potential. Our method is based on an apertureless single-mode-fiber confocal design. Our goal is to study a novel fiber-optic confocal approach for noninvasive ultrahigh-resolution three-dimensional bioimaging of cellular/intracellular and tissue samples beyond the diffraction barrier in the subwavelength (below 200 nm) nanometric range.
We have demonstrated the use of a fiber-optic based confocal microscope approach as an effective way to break the Rayleigh diffraction resolution barrier. To improve the dynamic range of the resolving laser power and to achieve an ultrahigh depth-resolution beyond the diffraction limit in the nanometric scale, we have designed a simple fiber-optic confocal microscope using a principal optical arrangement illustrated in (a) below. (b) shows a typical confocal response curve obtained using a fiber-optic confocal setup. In this case, we placed the sample slightly away from the focal point area to apply the DCM approach, so that its position is located in the middle area of the linear, sharp, diffraction-free slope on the left side of the axial response curve. In this way, the dynamic range and spatial sensitivity are significantly increased because the signal light that enters into the detecting fiber is highly sensitive to the sample position. As a result, we can get an ultrahigh depth-resolution beyond the diffraction limit in the nanometric range.
Below we illustrate experimental axially scanned confocal responses obtained with a fiber-optic confocal microscope when the DCM approach is applied using various sample axial displacements in the range of 2-100 nm. These results confirm the potential of the suggested fiber-optic confocal approach for getting a subwavelength depth resolution of 100 nm (a), 50 nm (Fb), 10 nm (c), and 2 nm (d), respectively, because the level of the registered signals is much higher than that of signal fluctuations observed.
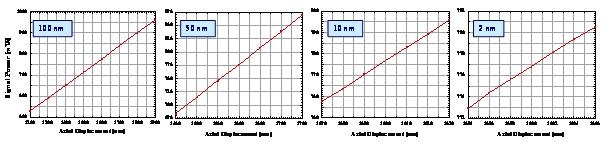
Experimental confocal responses obtained using the ultrahigh-resolution fiber-optic confocal microscope for achieving a subwavelength depth resolution of 100 nm (a), 50 nm (b), 10 nm (c), and 2 nm (d), respectively.
On a more theoretical note we have been looking at different ways to process an image to enhance resolution. In Small et al.'s paper we have shown that there is a very simple way to enhance our resolution down to 40% of the Rayleigh limit. This is achieved by looking at the shape of the profile generated by a source rather than its' peak value. We have shown that for discrete sources we may achieve a much higher than Rayleigh limited resolution with simple numerical processing. Further we showed that separation of near sources is also possible up to a certain limit. The numerical model for this approach is downloadable HERE.
 BACK TO TOP
BACK TO TOP