Research
The goal of our research is to understand the cellular and molecular mechanisms governing skeletal growth. This knowledge will then be applied to improve medical treatment in disorders of bone growth and in disorders of bone structure.
- Exome Sequencing to Identify Causes of Childhood Growth Disorders
- Genome-wide association studies (GWAS) to identify genes that control linear growth
- Targeted therapy for growth plate disorders
- Histone methyltransferases EZH1 and EZH2 regulate skeletal growth
- A neomorphic variant in transcription factor SP7 causes complex skeletal disease
- Loss-of-function variant in SPIN4 causes an X-linked overgrowth syndrome
- Linear growth disorders – a broader conceptual framework
- Fundamental limits on body growth
- How different bones in the body grow to different lengths
- A juvenile multi-organ growth-limiting program.
- Evolutionary conservation of the juvenile multi-organ growth-limiting program.
- Orchestration of the juvenile multi-organ growth-limiting program by E2F transcription factors.
- Growth-limitation: depletion of progenitor cells.
Exome Sequencing to Identify Causes of Childhood Growth Disorders
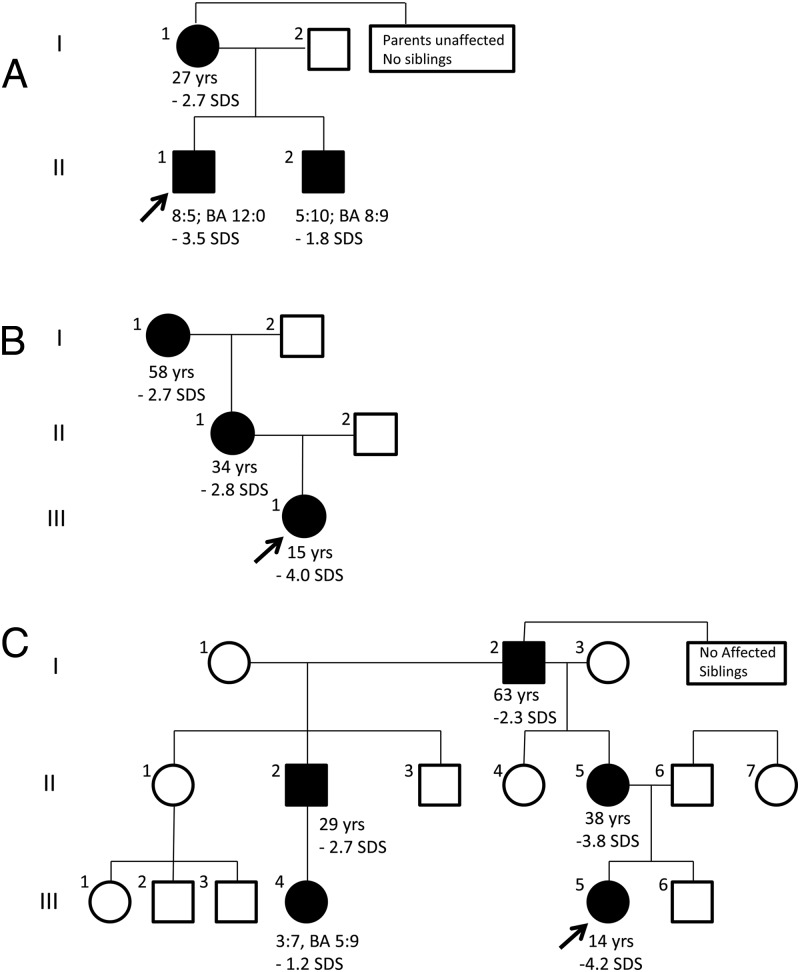
We use exome sequencing to identify monogenic causes of short stature. For example, we evaluated a boy with short stature (-3.5 SDS) and a bone age advanced 3 years compared to chronological age (Nilsson et al, 2014). His younger brother and mother were similarly affected. Exome sequencing identified a frameshift mutation (c.272delA) inACAN that co-segregated with the disorder and is extremely rare in unaffected individuals. Through a collaboration with colleagues at other institutions, we combined this family with two other families showing a similar phenotype and mutations in ACAN. Some affected individuals also manifested osteochondritis dissecans and early-onset osteoarthritis.ACAN encodes a proteoglycan that is an important component of cartilage extracellular matrix. It is expressed both in growth plate, providing an explanation for the short stature and advanced bone age, and also in articular and vertebral disc cartilage, providing an explanation for the joint disease. Three other families had been reported previously but with different clinical presentations – an evident skeletal dysplasia or early-onset joint disease. Our findings indicate that ACAN mutations, can present as autosomal-dominant short stature with advanced bone age and early growth cessation (Nilsson et al, 2014). Subsequent studies have further defined the phenotypic and genotypic spectrum of this disorder (Gkourogianni A et al. 2017).
Using similar approaches, we identified BRF1 mutations in a child with growth failure, markedly delayed bone age, and central nervous system anomalies (Jee et al. 2017), and identified QRICH1 mutations as a cause of a mild chondrodysplasia (Jee et al, 2019)
Genome-wide association studies (GWAS) to identify genes that control linear growth
Recently large meta-analyses of GWAS have identified >400 loci that are associated with adult height. Each locus implies the presence of a nearby gene that plays a biological role in human linear growth, but it is usually unclear which gene at each locus actually modulates height and which are innocent neighbors. To address this knowledge gap, we developed a new analytic approach to identify causative genes within these GWAS loci, based on the hypothesis that many of the causative genes within these loci influence height because they are expressed in and function in the growth plate. Therefore, we used expression microarray studies of mouse and rat growth plate, human disease databases, and a mouse knockout phenotype database to identify genes within the GWAS loci that are likely required for normal growth plate function. This analysis strongly implicated a large number of novel genes that regulate human growth plate chondrogenesis and thereby contribute to the normal variations in human adult height (Lui et al, 2012). Because polymorphisms in these genes modulate height within the normal range, we reason that more deleterious mutations in these same genes may cause pathological short stature. Therefore, the genes implicated by this approach are excellent candidates for causing skeletal dysplasias, syndromic short stature, and isolated short stature of unknown etiology. Subsequent collaborative studies have continued to use this approach to elucidate the genes that contribute to the genetic variation in stature (Pers et al, 2015), (Wood et al. 2014)
Targeted therapy for growth plate disorders
Many genetic disorders, including chondrodysplasias, and acquired disorders impair growth plate function, resulting in short and sometimes malformed bones. There are multiple endocrine and paracrine factors that promote chondrogenesis at the growth plate, which could potentially be used to treat these disorders. Targeting these growth factors specifically to the growth plate might augment the therapeutic skeletal effect while diminishing undesirable effects on non-target tissues. Using yeast display technology, we selected single-chain variable antibody fragments that bound to human and mouse matrilin-3, an extracellular matrix protein specifically expressed in cartilage tissue. We identified antibody fragments that bound matrilin-3 with high affinity and also bound with high tissue specificity to cartilage homogenates and to cartilage structures in mouse embryo sections. When injected intravenously in mice, the antibody fragments specifically homed to cartilage. Coupling these antibodies to chondrogenic endocrine and paracrine signaling molecules has the potential to open up new pharmacological approaches to treat childhood skeletal growth disorders (Cheung et al, 2015).
We next created fusion proteins of these cartilage-targeting single-chain human antibody fragment (CaAb) conjugated with insulin-like growth factor 1 (IGF-1), an endocrine and paracrine factor that positively regulates chondrogenesis. These CaAb-IGF-1 fusion proteins retained both cartilage binding and IGF-1 biological activity, and they were able to stimulate bone growth in an organ culture system. Using a GH-deficient (lit) mouse model, we found that subcutaneous injections of these CaAb-IGF-1 fusion proteins increased overall growth plate height without increasing proliferation in kidney cortical cells, suggesting on-target efficacy at the growth plate and less off-target effect on the kidney than IGF-1 alone. Alternate-day injections of these fusion proteins, unlike IGF-1 alone, were sufficient to produce a therapeutic effect. Our findings provide proof of principle that targeting therapeutics to growth plate cartilage can potentially improve treatment for childhood growth disorders (Lui et al, 2019) .
Histone methyltransferases EZH1 and EZH2 regulate skeletal growth
Histone methyltransferases EZH1 and EZH2 catalyse the trimethylation of histone H3 at lysine 27 (H3K27), which serves as an epigenetic signal for chromatin condensation and transcriptional repression. Genome-wide associated studies have implicated EZH2 in the control of height and mutations in EZH2 cause Weaver syndrome, which includes skeletal overgrowth. We found that the combined loss of Ezh1 and Ezh2 in chondrocytes severely impairs skeletal growth in mice. Both of the principal processes underlying growth plate chondrogenesis, chondrocyte proliferation and hypertrophy, are compromised. The decrease in chondrocyte proliferation is due in part to derepression of cyclin-dependent kinase inhibitors Ink4a/b, while ineffective chondrocyte hypertrophy is due to the suppression of IGF signalling by the increased expression of IGF-binding proteins. Collectively, our findings reveal a critical role for H3K27 methylation in the regulation of chondrocyte proliferation and hypertrophy in the growth plate, which are the central determinants of skeletal growth (Lui et al. 2016).
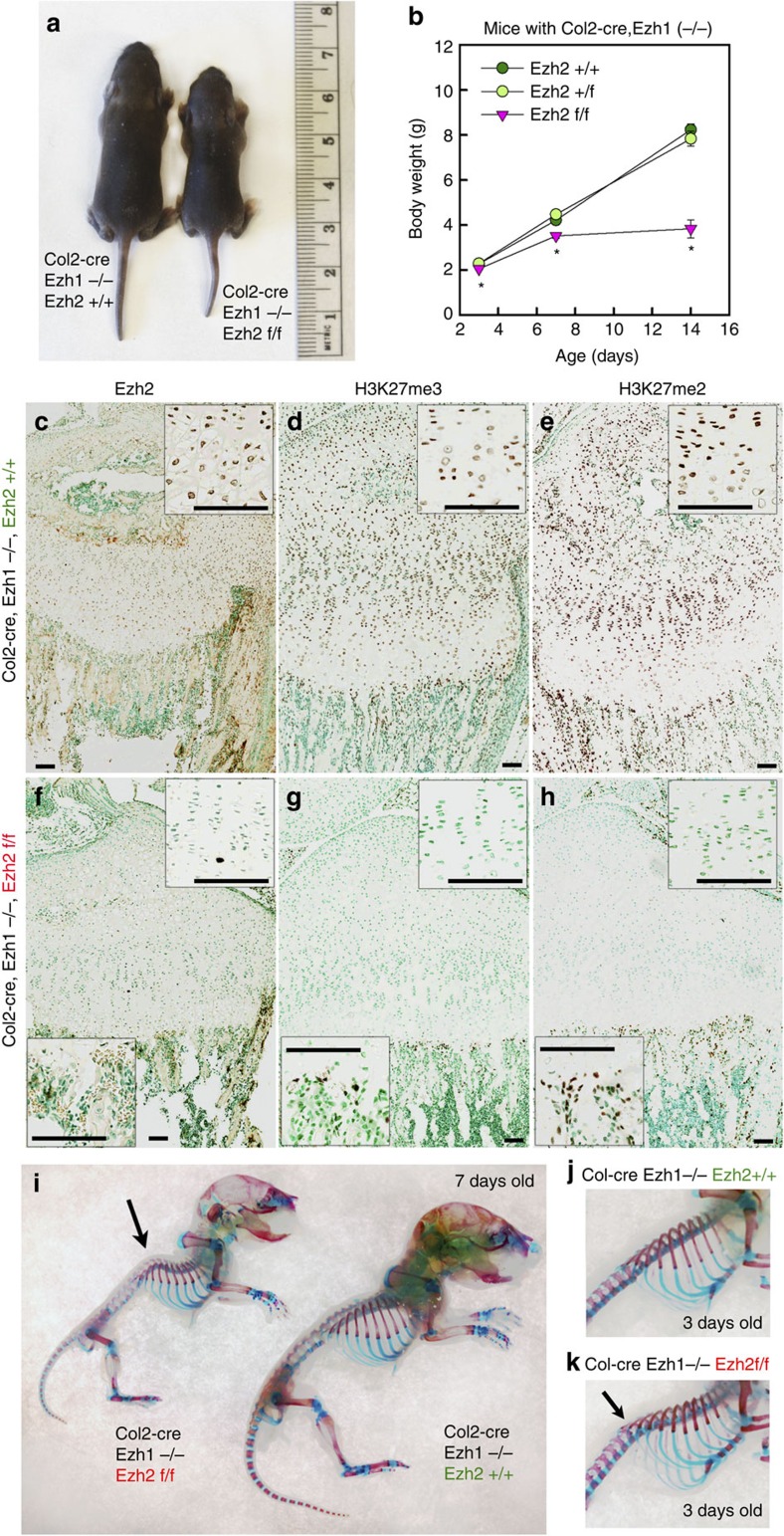
Exome sequencing was performed in a boy with tall stature, advanced bone age, and mild dysmorphic features. A de novo missense EZH2 mutation [c.1876G>A (p.Val626Met)] was identified. This finding was consistent with a recent report that mutations in EZH2 cause Weaver syndrome, which is characterized by tall stature, advanced bone age, characteristic facies, and variable intellectual disability. We next studied the mutation to determine whether Weaver syndrome is due to gain-of-function or loss-of-function mutations in EZH2. When expressed in growth plate chondrocytes, the mutant protein showed decreased histone methyltransferase activity. A mouse model carrying this EZH2 mutation was generated using CRISPR/Cas9. Homozygotes showed perinatal lethality, whereas heterozygotes were viable, fertile, and showed mild overgrowth. Thus, the mouse model with the same mutation as our patient recapitulated the Weaver overgrowth phenotype. Both homozygous and heterozygous embryos showed decreased H3K27 methylation. We concluded that EZH2 mutations found in Weaver syndrome cause a partial loss of function (Lui et al, 2018).
A neomorphic variant in transcription factor SP7 causes complex skeletal disease
We evaluated a boy who presented with multiple fragility fractures of long bones, heterogeneity of long bone cortical thickness, markedly thickened calvarium, craniosynostosis, scoliosis, and increased bone turnover. Exome sequencing identified a rare, de novo missense variant in SP7 (S309W). SP7 (also known as osterix) is a key transcription factor required for osteoblast differentiation.
To confirm that the variant caused the skeletal disease, we created heterozygous knock-in mice carrying the variant which showed a complex skeletal disorder similar to that of the human (see figure). In cell culture studies, we found that the SP7 variant produced an aberrant expression profile with accelerated osteoblast differentiation, but impaired bone matrix formation. Unlike other Sp family members, which directly bind a consensus GC box, Sp7 forms a complex with Dlx proteins to recognize AT-rich motifs. A gel shift assay showed that the variant increased binding to a GC-box but decreased binding to an AT-rich motif. ChIP-seq confirmed that the variant altered sequence-specific binding in the genome.
Thus, the findings reveal an unusual and interesting pathogenic mechanism. There have been many human diseases which have been found to be caused by loss-of-function variants in transcription factors and a smaller number due to gain-of-function variants. However, in this patient, the disease appears to arise because a neomorphic variant alters the DNA-binding specificity of a transcription factor, leading to aberrant regulation of gene expression (Lui et al, 2022).
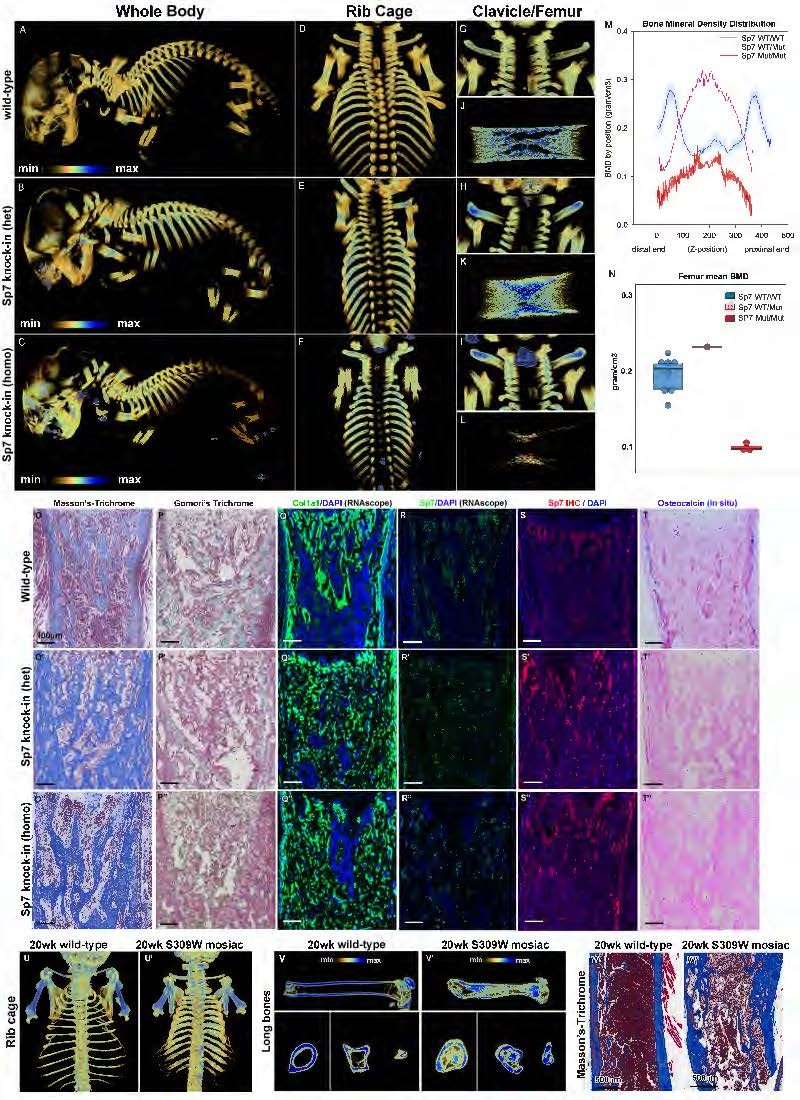
Figure. Mice with heterozygous S309W SP7 variant showed skeletal abnormalities. (A-L) micro-computed tomography analysis. Compared to newborn wild-type mice, both S309W SP7 heterozygous and homozygous knock-in mice showed increased bone thickness in the ribs (D-F) and clavicles (G-I). Long bones in S309W knock-in mice were shortened but increased in diameter (J-L), with redistribution of bone mineralization. In the heterozygous knock-in mice, trabecular bone density was decreased at the metaphyses but increased at the diaphyses of the femurs. Homozygous knock-in mice showed a similar pattern but decreased overall bone density (M,N). (O-T) Histology in the proximal tibial metaphysis showed markedly increased extracellular matrix production in the mutants (O-O”) but decreased mineralized bone (P,P’). Col1a1 (Q-Q”) and Sp7 (R-R”, S-S”) expression occurred more sporadically and in a disorganized pattern in the mutants and osteocalcin mRNA expression was diminished in the cortex (T-T”). (U-W) The S309W mosaic mouse analyzed by micro-CT imaging at 20 weeks of age (compared to age-matched wild-type). The mosaic mouse similarly showed increased bone thickness in the ribs and clavicles (U), shortened but widened long bone with abnormal medullary cavity (V). Histology sections showed uneven cortical thickness (W).
Loss-of-function variant in SPIN4 causes an X-linked overgrowth syndrome
We recently studied a child with generalized overgrowth of prenatal onset (see figure below).
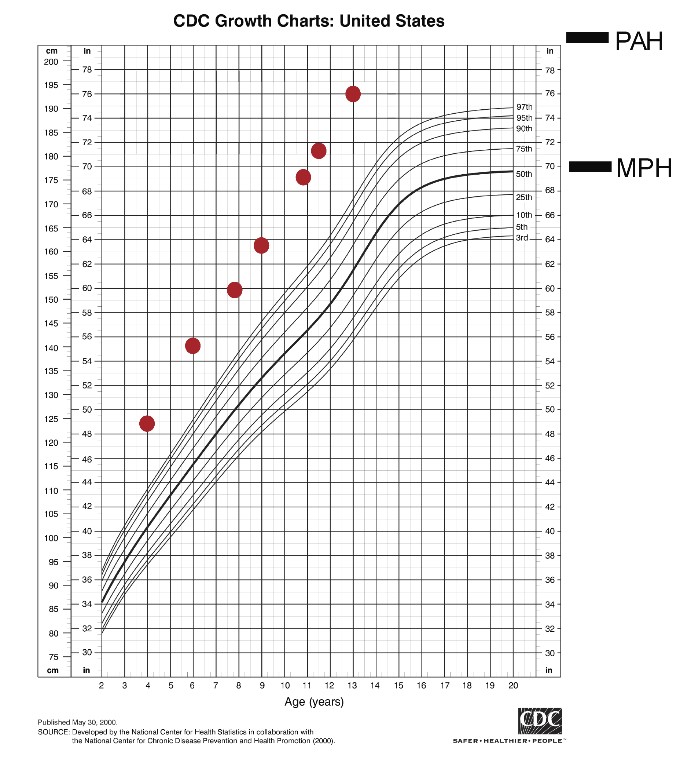
Figure. Growth chart of a child with overgrowth caused by a variant in SPIN4.
Exome sequencing identified a hemizygous frameshift variant in Spindlin 4 (SPIN4), with X-linked inheritance. Ablation of Spin4 in mice recapitulated the human phenotype with generalized overgrowth, including increased longitudinal bone growth (see figure below).
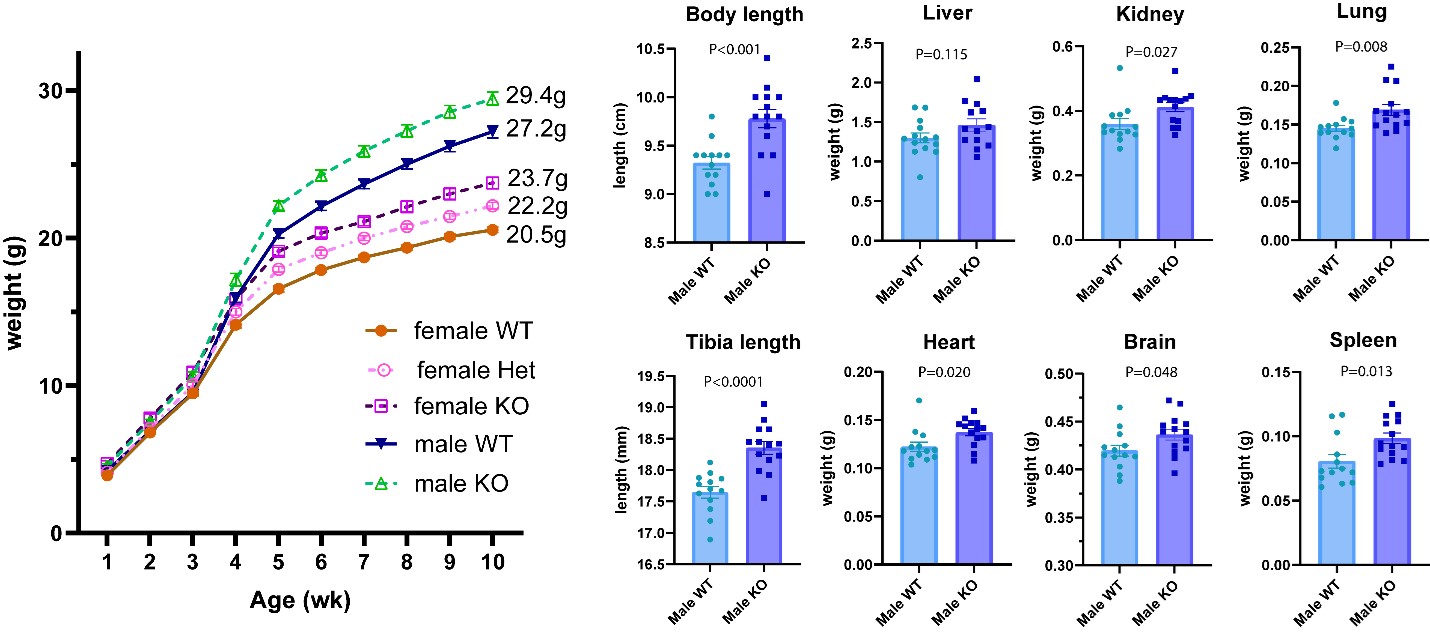
Figure. Body weight (left) and sizes of multiple organs (right) are increased in mice lacking SPIN4
We found evidence that SPIN4 binds specific histone modifications (see figure below), promotes canonical WNT signaling, and inhibits cell proliferation in vitro and that the identified frameshift variant had lost all of these functions.
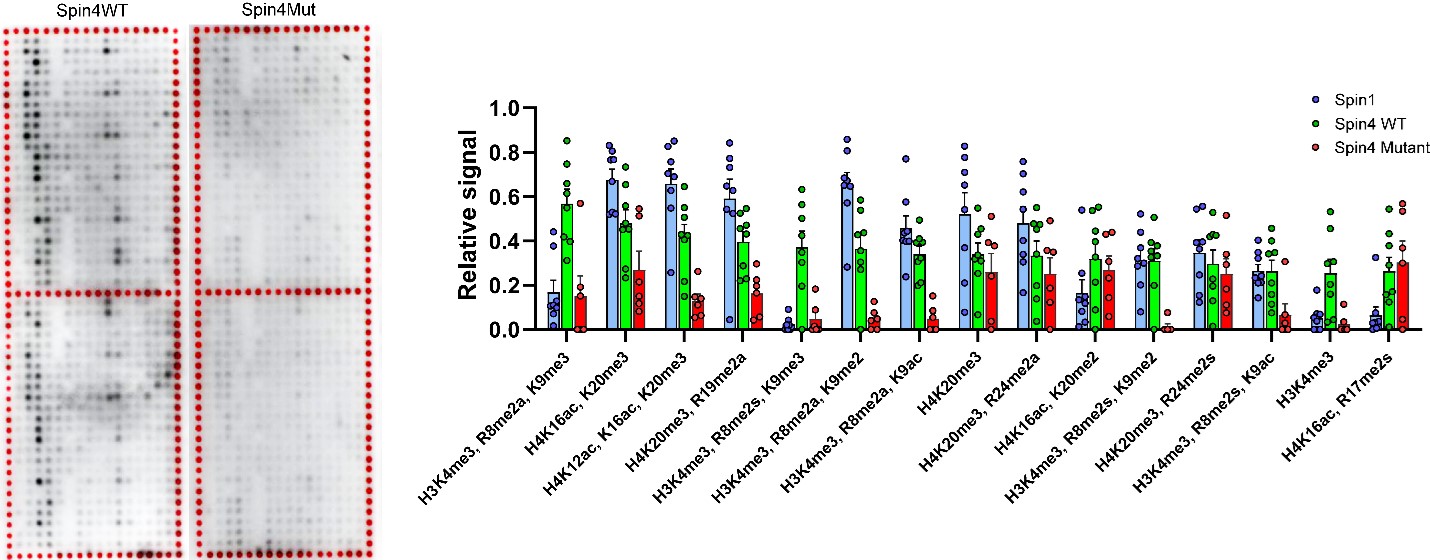
Figure. Spin4 binds to specific modified histones. Histone peptide arrays were used to assess histone binding properties of wild-type (WT) and mutant (Mut) SPIN4. Each spot on the array contains a peptide portion of a histone that has undergone specific posttranslational modifications. Darker color indicates greater binding of the indicated Spin protein to that modified peptide. WT SPIN4 binds to specific modified histone peptides, while Mut SPIN4 showed substantially diminished binding. The box-and-whisker plot shows these data quantitatively.
Growth plate analysis revealed increased cell proliferation in the proliferative zone and an increased number of progenitor chondrocytes in the resting zone. We also found evidence of decreased canonical Wnt signaling in growth plate chondrocytes, providing a potential explanation for the increased number of resting zone chondrocytes. Taken together, our findings provide strong evidence that SPIN4 is an epigenetic reader that negatively regulates mammalian body growth, and that loss of SPIN4 causes an overgrowth syndrome in humans, expanding our knowledge of the epigenetic regulation of human growth.
Linear growth disorders – a broader conceptual framework
Recently many new important systems that regulate childhood linear growth have been identified, most of which operate locally in the growth plate. Mutations in the genes that encode many of these local proteins, e.g. NPR2, FGFR3, ACAN, PTHrP, cause short stature or tall stature. Evidence, to which we contributed, has also emerged that height variation within the normal range is due in large part to variants in these local regulatory systems.
To make sense out of all these exciting new findings and to incorporate them into our clinical thinking as pediatric endocrinologists, our group has advocated adopting a broad conceptual framework based on growth plate biology. We have argued that it is very helpful to think about short and tall stature in terms of growth plate chondrogenesis, the fundamental process that drives linear growth in children. Using this framework, we can conceptualize all the myriad of genetic defects that affect linear growth in terms of the mechanisms by which they affect chondrogenesis. We have explained this conceptual framework in two recent reviews, “Short and Tall Stature: a New Paradigm Emerges,” in Nature Reviews Endocrinology (Baron et al. 2015) and “The Biology of Stature,” in The Journal of Pediatrics (Jee et al. 2016).
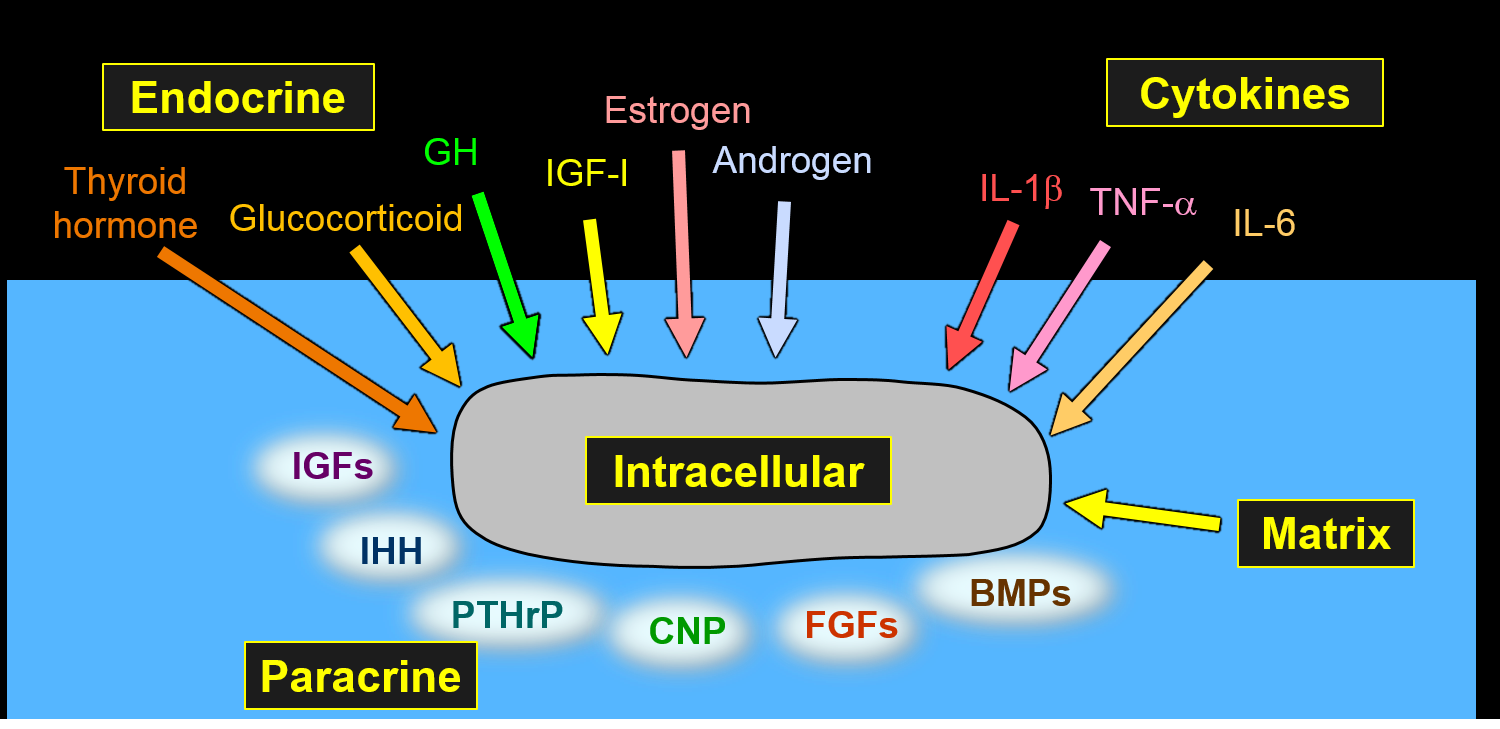
Fundamental limits on body growth
One of the remaining fundamental mysteries in biology is how organs and organisms know when to stop growing. In mammals, by the time of adulthood, body growth has ceased or slowed dramatically. Importantly, this cessation or near cessation of growth does not occur abruptly, but rather is the end result of a progressive decline in growth rate that begins in early life. This growth deceleration occurs not only in linear growth, which reflects growth of chondrocytes in the growth plate, but also in growth of other tissues and organs, and consequently in growth of overall body mass. Growth deceleration results primarily from a decline in the rate of cell proliferation but the underlying mechanisms that suppress proliferation remain poorly understood. Understanding the fundamental mechanisms that cause growth deceleration and thus govern adult body size is likely to yield insights into the many human genetic disorders that cause childhood growth failure and overgrowth, many of which remain poorly understood. Because body growth limitation involves complex, potent mechanisms to suppress cell proliferation, disruption of these mechanisms may contribute to oncogenesis. Additionally, elucidating the physiological mechanisms that normally suppress proliferation in juvenile life might suggest new approaches to achieve therapeutic tissue regeneration, either in vivo or ex vivo.
How different bones in the body grow to different lengths
Bones at different anatomical locations vary dramatically in size. For example, human femurs are 20-fold longer than the phalanges in the fingers and toes. The mechanisms responsible for these size differences are poorly understood. Bone elongation occurs at the growth plates and advances rapidly in early life but then progressively slows due to a developmental program termed "growth plate senescence." This developmental program includes declines in cell proliferation and hypertrophy, depletion of cells in all growth plate zones, and extensive underlying changes in the expression of growth-regulating genes. Here, we show evidence that these functional, structural, and molecular senescent changes occur earlier in the growth plates of smaller bones (metacarpals, phalanges) than in the growth plates of larger bones (femurs, tibias) and that this differential aging contributes to the disparities in bone length. We also show evidence that the molecular mechanisms that underlie the differential aging between different bones involve modulation of critical paracrine regulatory pathways, including insulin-like growth factor (Igf), bone morphogenetic protein (Bmp), and Wingless and Int-1 (Wnt) signaling. Taken together, the findings reveal that the striking disparities in the lengths of different bones, which characterize normal mammalian skeletal proportions, is achieved in part by modulating the progression of growth plate senescence (Lui et al, 2018).
A juvenile multi-organ growth-limiting program.
We identified a complex growth-limiting genetic program that drives this growth deceleration during juvenile life ( Lui et al, 2010). It occurs in multiple tissues and thus explains the concurrent growth deceleration that occurs in various organs. This shared program involves the down-regulation of hundreds of growth-promoting genes with age and is driven not by time but rather by growth itself (Lui et al, 2010) .
Evolutionary conservation of the juvenile multi-organ growth-limiting program.
We also found that this growth-limiting genetic program was conserved among mammals but that its pace has been evolutionarily modulated to achieve species differences in body size. Mammalian body size varies enormously among species, from the Etruscan shrew, with an adult body mass <2 g to the blue whale, with adult body mass >100,000 kg. This wide variation in size is due in part to differences in the duration of body growth; somatic growth in small mammals generally lasts for weeks, whereas large mammals continue to grow for years. We found evidence that the greater duration of growth in larger mammals occurs because the growth-regulating genetic program plays out more slowly, with a more gradual decline in the expression of growth-promoting genes (. Our findings are consistent with the hypothesis that the juvenile growth-regulating genetic program proceeds at a slower pace in larger mammals, allowing for more prolonged growth and greater adult body size (Delaney et al, 2014).
Orchestration of the juvenile multi-organ growth-limiting program by E2F transcription factors.
We have also explored the molecular mechanisms that orchestrate this growth-limiting genetic programing, driving downregulation of hundreds of growth-promoting genes in multiple tissues. We found evidence that E2F transcription factor 3 (E2F3) acts as a molecular master switch to help drive these changes in expression (Lui et al, 2013). We showed that E2F3 strongly promotes expression of many genes in this program. The expression of E2F3 itself declines with age postnatally in multiple tissues and this decline then drives decline of many genes in the growth-limiting program. To explore the mechanisms by which E2F3 achieves this orchestration, we focused on IGF2, one of the many growth-promoting genes that are downregulated with age. We found that E2F3 regulates IGF2 expression by a binding to E2F binding sites in the IGF2 promoter region and affecting transcription. We also found evidence that this regulation of IGF2 by E2F3 occurs not only in normal juvenile tissues, regulating physiological growth deceleration but also in malignancies; E2F3 overexpression in human cancers induces IGF2 overexpression, likely contributing to rapid proliferation of these malignancies (Lui et al, 2013).
Growth-limitation: depletion of progenitor cells.
Growth deceleration that occurs in the growth plate, and consequently in linear growth, involves molecular mechanisms similar to those in non-skeletal tissues. Study of these mechanisms in growth plate cartilage has an advantage in that the progenitor cells, the rapidly proliferating cells and the terminally differentiated cells are spatially compartmentalized and therefore each can be studied separately. With age, growth plate cartilage undergoes programmed senescence, eventually causing cessation of bone elongation and epiphyseal fusion. We previously showed that estrogen accelerates this developmental process (Weise et al, 2001). Recently, we explored the underlying mechanisms and found evidence that senescence occurs because progenitor cells in the resting zone are depleted in number and that estrogen acts by accelerating this depletion (Nilsson et al, 2014). These findings provide a mechanistic understanding of common clinical phenomena, including the accelerated loss of growth potential in children with precocious puberty and the delayed loss of growth potential in individuals with mutations in aromatase or the estrogen receptor.
 BACK TO TOP
BACK TO TOP