Research
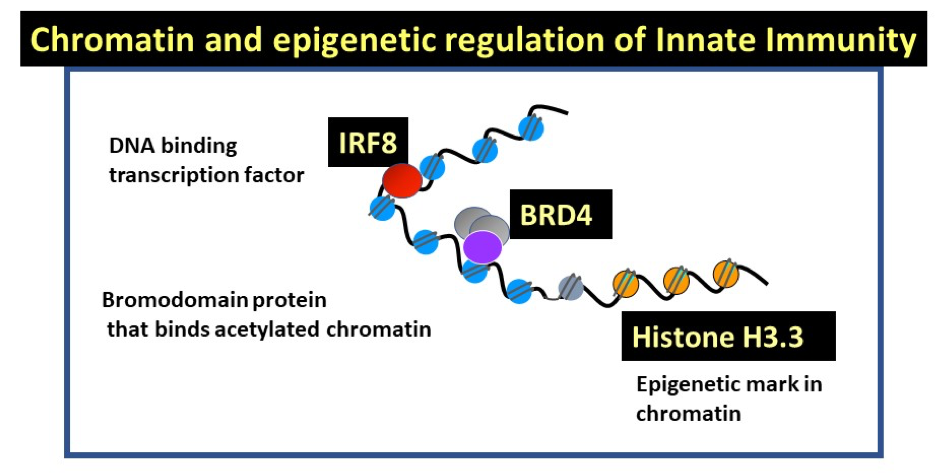
Keiko Ozato’s laboratory studies chromatin and epigenetic regulation of innate immunity. They work on three factors that control essential aspects of innate immunity, i.e., natural resistance against infection and accompanying inflammatory stress response (Figure 1). IRF8 is a sequence specific transcription factor that activates both anti-microbial resistance and inflammatory reactions. IRF8 acts primarily in myeloid cells, macrophages, dendritic cells (DCs) and microglia. BRD4 is a bromodomain containing nuclear factor that binds to acetylated histones. It also promotes transcriptional elongation of many cellular and viral genes. Small molecule inhibitors that block binding to acetylated histones block growth of many blood cancers, which has opened new therapeutic strategy. The histone H3.3, variant of H3, also known as replacement histone, is deposited in actively transcribed genes concomitant with transcription. The presence of H3.3 signifies prior transcription and is the hallmark of epigenetic regulation.
Click image to view.
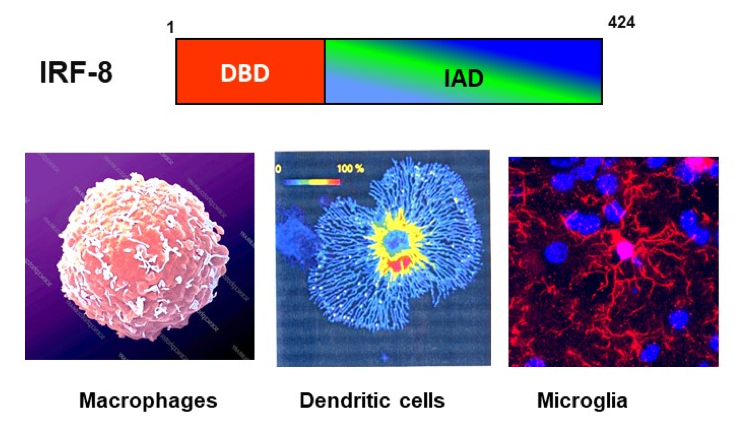
IRF8, critical for innate immunity (Figure 2)
- IRF8 regulates development of myeloid lineage cells: It is expressed in a series of myeloid progenitors in bone marrow that are derived from hematopoietic stem cells. It promotes LY6C+ monocyte macrophage lineage, while suppressing neutrophil pathways. IRF8 is also important for development of two types of DCs, CD8+ DCs and plasmacytoid DCs. Conversely, it inhibits osteoclast development, thus playing a role in bone homeostasis. IRF8 binds to specific DNA motifs, ISRE and IRF-ETS composite by interacting partner factors, SPI1 (PU.1) and other IRF members. IRF8 occupies distant enhancers that support expression of genes that help define myeloid cell identity.
- IRF8 is a tumor suppressor and inhibits growth of various leukemia. Irf8-/- (IRF8 KO) mice present chronic myeloid leukemia like syndrome and produce excessive amounts of immature neutrophils. In humans IRF8 is downregulated in a variety of myeloid cells in leukemia patients. While IRF8 interferes with the BCR/ABL pathway in some leukemia, the precise mechanisms by which IRF8 controls leukemia growth is not fully understood.
- IRF8 confers innate resistance to infection through many varied mechanisms. It stimulates production of interferons (Type I, IFNg and IFN l, thus inducing many IFN stimulated genes (ISGs). IRF8 induces reactive oxygen species, many cytokines and chemokines involved in pathogen control. IRF8 stimulates phagocytosis, activates autophagy and enhances lysosomal functions to degrade ingested pathogen components. In contrast to the important host protection against pathogens, IRF8 contributes to aggravation of autoimmune diseases such as experimental allergic encephalopathy (a mode for MS) and systemic lupus erythematosus.
Ongoing Research: Recently, we began studying IRF8 in microglia and its role in neuroinflammation. IRF8 and SPI1 (PU.1) play a central role in early microglia development in the embryonic stage. In adult brain, IRF8 KO microglia, although present in normal number, are abnormal in morphology, deficient in cytoplasmic extensions. Further microglia specific surface markers, such as Iba1 and Cx3cr1 are lower in expression. RNA-seq data revealed that IRF8KO microglia are deficient in other genes that define microglia, such as P2ry12, Sal I, Sparc and Pilra/b. Moreover, IRF8KO microglia expressed many so called “disease-associated microglia (DAM)” genes found in Alzheimer’s disease (AD) and its mouse models, including Apoe, Cst1, Cst7. Another unusual feature of IRF8 KO microglia is the expression of some ISGs including Irf4 and Irf7. These results indicate that IRF8 controls microglia gene expression programs in adult brain, and broadly influences microglia’s functionality. Without IRF8, microglia go awry in their trait and behavior. Based on these data, IRF8 is likely to have dual roles and affects the course of neuroinflammation positively and negatively, depending on the types and mechanisms of inflammation. For example, Trex-1 KO mice, when crossed withIRF8 KO mice gained longer survival time. On the other hand, IRF8 KO microglia did not fully establish LPS induced tolerance, relative to wild type (WT) microglia.
Click image to view.
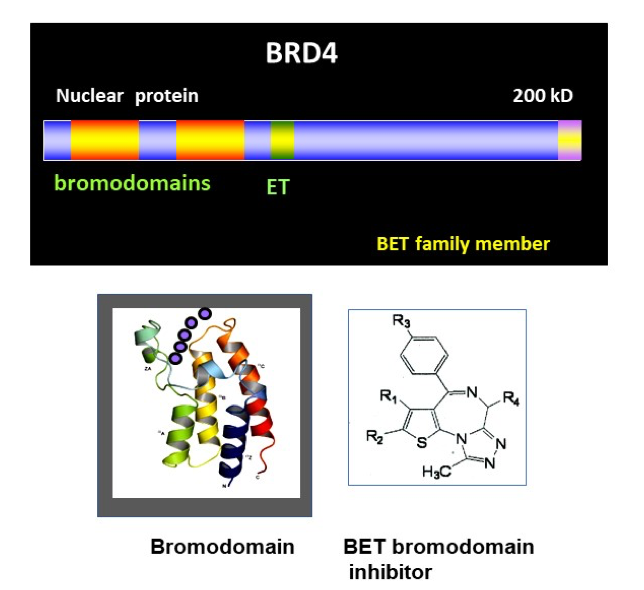
BRD4, chromatin reader that controls transcription (Figure 3)
- We isolated the murine Brd4 gene and reported about it in 2000. BRD4 carries two bromodomains that bind to acetylated histone tails. While bromodomains are present in many other chromatin binding factors such as histone acetylases, BRD4 belongs to a small, BET subfamily that shares greater structural similarity than other proteins. BRD4 also has a unique C-terminal motif, through which it binds to transcription elongation factor P-TEFb and stimulates transcription of many cellular and viral genes. BRD4 is essential for mammalian development, as standard knockout is early embryonic lethal in the mouse. Since then, small molecule inhibitors have been developed that block binding of BET bromodomains to acetylated histone tails. These inhibitors arrest growth of various leukemia including acute and chronic myeloid leukemia, lymphoma and myeloma and some solid cancers. Some of new generation BET small molecule inhibitors are tested in the clinical setting. BET inhibitors, along with other drugs that target various histone modifications have opened a new line of therapeutic approach where epigenetic traits can be modified.
Ongoing research: The dramatic effects of BET inhibitors on arresting blood cancer growth made it important to clarify what function BRD4 has in normal cells. To this end, we constructed Brd4f/f mice and knocked out Brd4 during hematopoiesis and differentiated macrophages. Our results demonstrated that BRD4 is obligatory for the development of embryonic and adult hematopoietic stem cells, which require proliferation. BRD4 was found to be critical for proliferation of macrophages and thymic double negative cells, although the lack of BRD4 only partially interfered differentiation. These results are consistent with the role of BRD4 for general cell cycle progression. To further probe the mechanisms by which BRD4 regulates cell cycle, we synchronized Brd4 KO and WT mouse embryonic fibroblasts by serum starvation and compared cell cycle progression after release. Results revealed that Brd4 deletion inhibits transition from G0 to G1, G1 to S and S to G2/M, indicative of compound defects. Furthermore, a considerable fraction of Brd4 KO cells failed to achieve mitosis. Gene profiling data showed that many cell cycle regulated genes were greatly downregulated in BRD4 KO cells found for genes specific to each stages of cell cycle. ChIP-seq analysis revealed that BRD4 occupies numerous cell cycle regulated genes in a constitutive manner, although BRD4 peaks rose dramatically during the time of expression for many of these genes. These results lead us to conclude that BRD4 plays an essential role in promoting cell cycle progression in normal cells, not limited to cancer cells. These results suggest that the use of BET inhibitors needs calibration to minimize adverse effects on normal cells.
Our separate ChIP-seq analysis of BM macrophages demonstrated that BRD4 is a major constituents of super-enhancers. Super-enhancers are a large stretch of regulatory sequences that is densely occupied by chromatin- and transcriptional-regulators, RNA polymerase II, with the H3K2ac histone mark. Strikingly. LPS stimulation dissolved many of super-enhancers present in resting macrophages almost instantly. LPS treatment led to the formation of a new set of super-enhancers. Many of these new super-enhancers were occupied by BRD4, and found near the genes induced by LPS, such as Tnf, Il1b and ISGs. These data illustrate broad and varying roles that BRD4 plays in regulating macrophage inflammatory processes. An unexpected observation was that NF-kB occupancy was higher in BRD4 KO macrophages found in many proinflammatory genes. Consequently, these highly inflammatory genes were expressed at higher levels in BRD4 KO macrophages than WT counterparts. Our data point to a novel anti-inflammatory role for BRD4.
Click image to view.
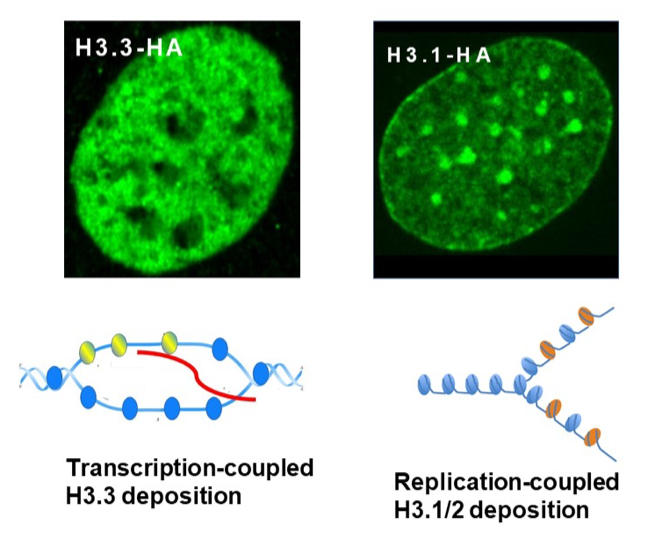
Histone H3.3 and epigenetic marking (Figure 4)
- IFN induced H3.3 gene marking. Our previous observation that upon IFN treatment, H3.3 accumulates on ISGs n in mouse embryonic fibroblasts (MEFs) has become our model for H3.3 deposition as a function of gene marking. This deposition was concurrent with transcriptional elongation of ISGs. IFN induced H3.3 deposition has been confirmed in three independent means; a) cells exogenously expressing HA-tagged H3.3, b) cells in which exogenous H3.3 is transiently activated by the SNAP-technology, and c) cells in which endogenous H3.3 genes (H3f3a, H3f3b) were replaced by HA-tagged H3.3 gene by homologous recombination. The initial finding was extended to genome-wide ChIP-seq which also confirmed IFN stimulated H3.3 deposition. As expected, H3.3 deposition was found in many constitutively expressed genes irrespective of IFN stimulation. The amount of H3.3 deposition in constitutively expressed genes roughly correlated with RNA transcript levels.
- H3.3 marking and transcriptional memory. In fibroblasts, about half of ISGs were without H3.3 before IFN treatment, and gained H3.3 marking after stimulation. However, the rest of ISGs were already marked with H3.3, even before IFN stimulation. Significantly, those ISGs showing new H3.3 deposition exhibited a clear memory response, in that these ISGs were induced more robustly when stimulated by the second IFN treatment. ISG induction occurred faster and at higher levels. Thus, H3.3 deposition was concurrent with the establishment of epigenetic memory. Although suggestively, it is not clear at present whether H3.3 deposition is responsible for memory.
Ongoing research: The histone chaperon, HIRA mediates deposition of histone H3.3 in actively expressed genes, whereas ATRX/DAXX mediates H3.3 deposition in pericentric heterochromatin and telomere. We constructed Hiraf/f mice and investigated the consequence of HIRA gene loss in hematopoietic stem cells (HSCs). We found that Hira KO mice are born at normal Mendelian ratio and reach maturity, but suffer drastic depletion of all types of blood cells, from platelets to erythrocytes, to lymphocytes and to a lesser degree myeloid cells. The lack of blood cells was attributed to a sharp reduction of HSCs in bone marrow (BM), cells with LSK markers. As a result, multipotent pluripotent progenitor cells were also sharply reduced. Adoptive transfer experiments showed that the lack of HSCs is due to the inability of Hira KO mice to generate and maintain BM HSCs, rather than a defect in the BM environment. We found, by RNA-seq analysis of BM HSCs (LSK) that a large number of transcription factors that define HSCs such as Meis, Jun, Fos, Egr1,Mecom were strongly downregulated in Hira KO HSCs relative to WT HSCs. We carried out ATC-seq analyses to find out genome-wide chromatin accessibility sites. Results showed that many ATAC-seq peaks present in WT HSCs were lost in Hira KO HSCs. Even more interestingly, many of lost peaks were near or at the genes involved in shaping HSCs. Our results illustrate that HIRA plays a critical role in setting he chromatin environment conferring HSC identity. Our result are interesting in light of the fact that the HIRA gene is deleted in some of DiGeorge syndrome patients. These patients present complex heterogenous disease phenotypes including anemia and immunodeficiency. Our data suggest that some of DiGeoroge syndrome phenotypes may be caused by the absence of HIRA.
We have recently constructed H3.3 conditional knockout mice, lacking both H3.3A and H3.3B in myeloid cells. These mice are derived from H3.3af/f and H3.3bf/f mice. Compared to Hira KO mice, H3.3 KO cells are expected to have more extensive defects, since ATRAX mediated H3.3 deposition is also abrogated in these mice.
Click image to view.
 BACK TO TOP
BACK TO TOP