Research
Our laboratory investigates the molecular mechanisms by which transmembrane proteins (referred to as “cargo”) are sorted to different compartments of the endomembrane system in eukaryotic cells. This system comprises an array of membrane-enclosed organelles including the endoplasmic reticulum (ER), the Golgi apparatus, the trans-Golgi network (TGN), endosomes, lysosomes, lysosome-related organelles (LROs) (e.g., melanosomes and platelet dense bodies), and different domains of the plasma membrane in polarized cells (e.g., epithelial cells and neurons) (Figure 1). Transport of cargo between these compartments is mediated by carrier vesicles or tubules that bud from a donor compartment, translocate through the cytoplasm, and eventually fuse with an acceptor compartment. Work in our laboratory focuses on the molecular machineries that mediate these processes, including (1) sorting signals and adaptor proteins that select cargo proteins for packaging into the transport carriers, (2) microtubule motors and their cognate adaptors that drive movement of the transport carriers and other organelles through the cytoplasm, and (3) tethering factors that promote fusion of the transport carriers to acceptor compartments. We study these machineries in the context of different intracellular transport pathways, including endocytosis, recycling to the plasma membrane, retrograde transport from endosomes to the TGN, biogenesis of lysosomes and LROs, and polarized sorting in epithelial cells and neurons. Knowledge gained from this basic research is applied to the elucidation of disease mechanisms, including congenital disorders of protein traffic such as the pigmentation and bleeding disorder Hermansky-Pudlak syndrome (HPS) and various neurodevelopmental and neurodegenerative disorders (e.g., hereditary spastic paraplegias and pontocerebellar hypoplasias). The sections below summarize some of our recent findings in these projects.
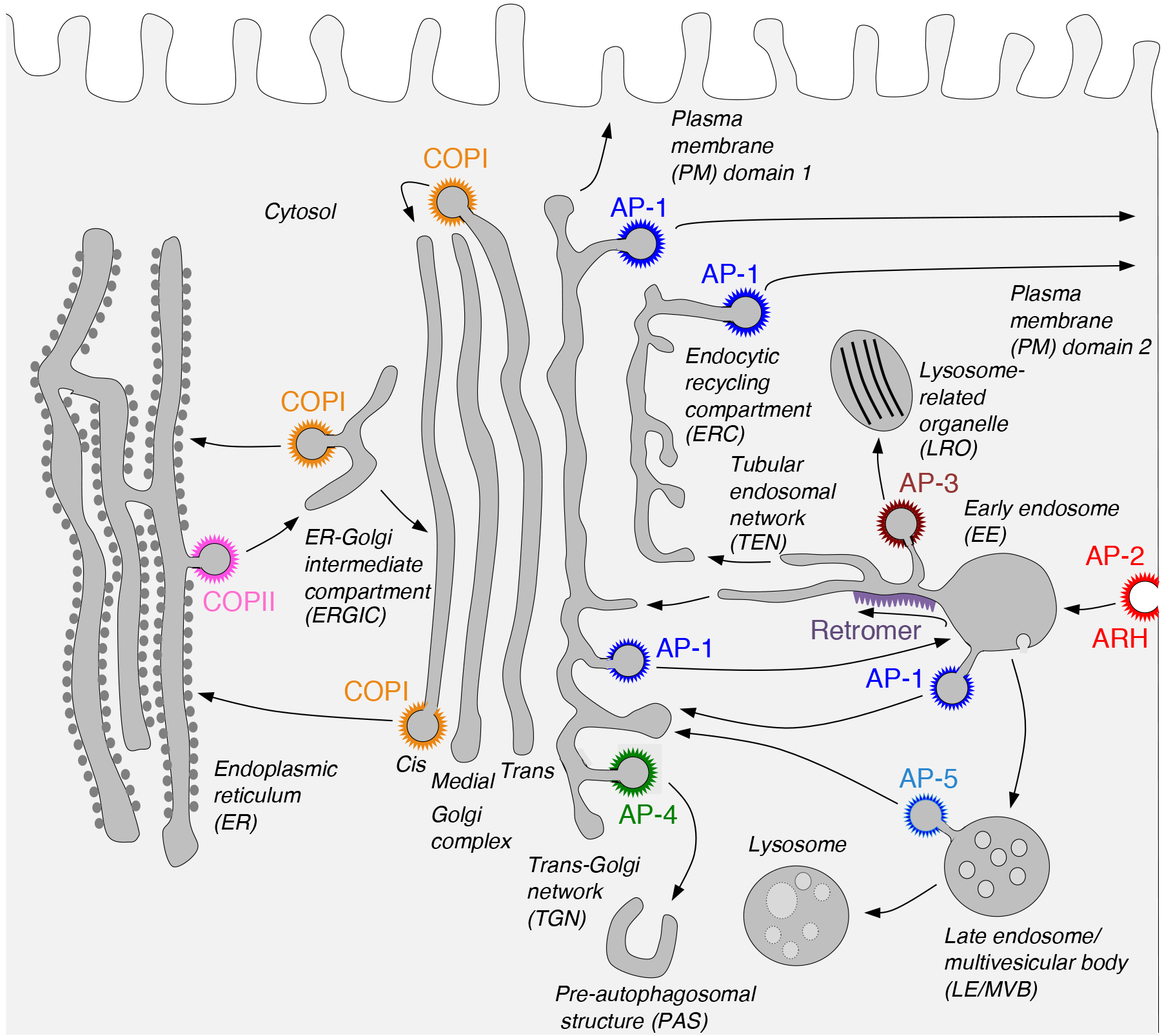
Click image to enlarge.
AP-4 promotes export of the autophagy protein ATG9A from the TGN: insights into the pathogenesis of hereditary spastic paraplegia caused by mutations in AP-4 subunit genes
A major focus of research in our laboratory is on processes mediated by recognition of sorting signals in the cytosolic tails of transmembrane proteins by adaptor proteins that are components of protein coats (Fig. 1). Two types of sorting signals known as “tyrosine-based” and “dileucine-based” participate in various sorting events, including endocytosis, transport to lysosomes and melanosomes, and polarized sorting to the basolateral surface of epithelial cells and the somatodendritic domain of neurons (Dell'Angelica and Bonifacino. Annu Rev Cell Dev Biol. 2019. 35:131-168). In previous work, we showed that tyrosine-based signals bind to a conserved site on the mu1, mu2 and mu3 subunits of three hetero-tetrameric adaptor protein (AP) complexes, AP-1, AP-2 and AP-3, respectively. Dileucine-based signals, on the other hand, bind to a different site on the surface of two subunits, gamma-sigma1, alpha-sigma2 and delta-sigma3, of the corresponding AP-1, AP-2 and AP-3 complexes. We subsequently found that a subset of tyrosine-based sorting signals fitting the consensus motif YXX[FL]E (where X is any amino acid) bind to the mu4 subunit of another AP complex named AP-4. This complex localizes to the TGN, but until recently its function was unknown. We recently discovered that AP-4 recognizes a YXX[FL]E-type motif (YQRLE) in the cytosolic tail of ATG9A, the only transmembrane component of the core autophagy machinery (Mattera et al. Proc Natl Acad Sci U S A. 2017. 114:E10697-E10706; De Pace et al. PLoS Genet. 2018. 14:e1007363). This recognition promotes export of ATG9A from the TGN to pre-autophagosomal structures (PAS), where ATG9A contributes to the initiation and expansion of autophagosomes. Inactivating mutations in AP-4 subunit genes result in ATG9A accumulation in the TGN (Fig. 2), with consequent autophagic defects. These defects likely underlie the pathogenesis of a subset of hereditary spastic paraplegias (i.e., SPG47, SPG50, SPG51 and SPG52) caused by mutations in AP-4 subunit genes and thus referred to as “AP-4-deficiency syndrome”. This syndrome is one of many diseases, known as “coatopathies”, which are caused by mutations in components of protein coats (Dell'Angelica and Bonifacino. Annu Rev Cell Dev Biol. 2019. 35:131-168). The elucidation of the cellular role of AP-4 should contribute to the development of new therapeutic strategies for this form of hereditary spastic paraplegia.
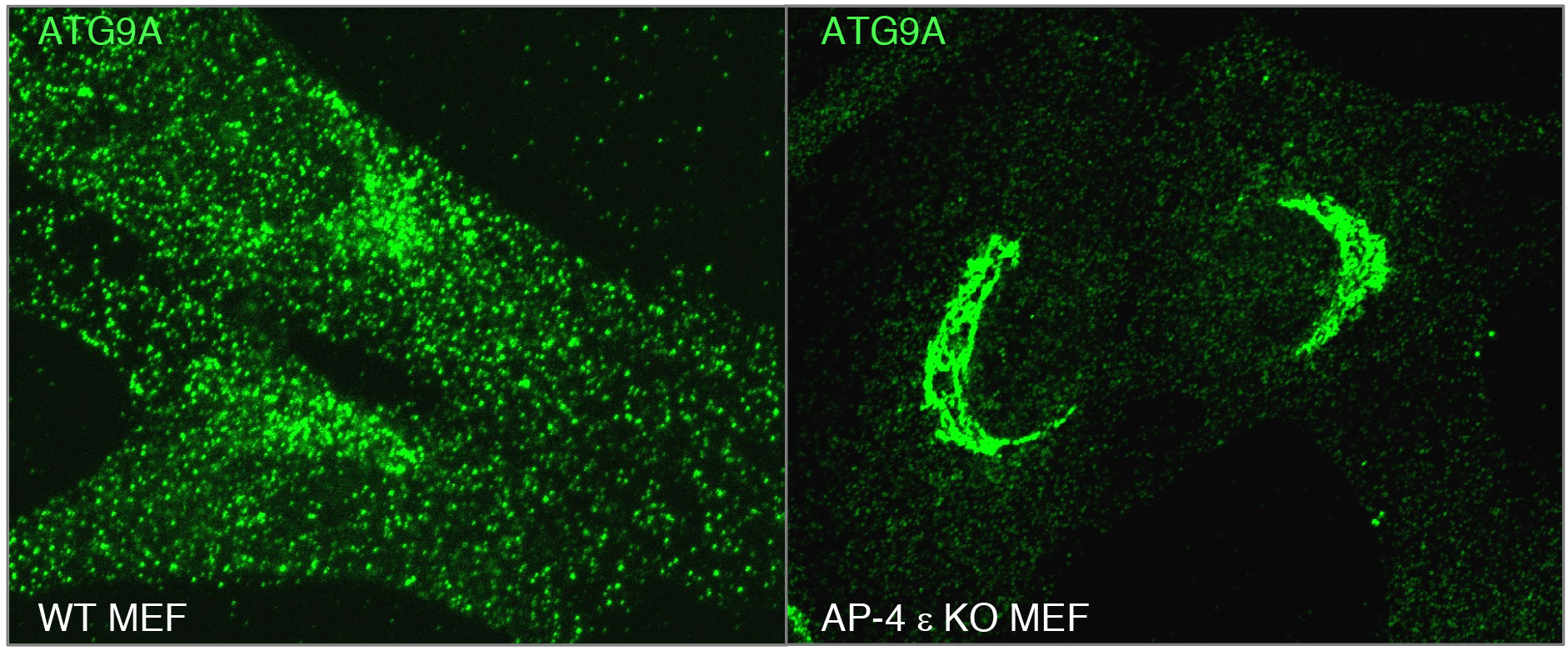
Click image to enlarge.
Mechanisms and functions of lysosome positioning
Another line of research in the laboratory focuses on the molecular mechanisms of organelle positioning and motility within the cytoplasm. This research started with our discovery of a hetero-octameric complex named BORC that regulates lysosome positioning (Pu et al. Dev Cell. 2015. 33:176-88; Guardia et al., Cell Rep. 2016. 17:1950-1961). We found that BORC associates with lysosomes, where it functions to recruit the small GTPase ARL8. This initiates a chain of interactions that couple lysosomes to kinesin-1 and kinesin-3, leading to lysosome movement toward the cell periphery (Fig. 3). Mutations in BORC cause collapse of the lysosomal population into the pericentrosomal area (Pu et al. Dev Cell. 2015. 33:176-88), and are the cause of another type of hereditary spastic paraplegia (Charng et al. BMC Med Genomics. 2016. 9:42). Further work showed that BORC- and ARL8-dependent lysosome positioning influences the function of lysosomes in various cellular processes, including autophagy, cell adhesion, cell migration, and metabolic signaling (Pu et al. Dev Cell. 2015. 33:176-88; Jia et al. Autophagy. 2017. 13:1648-1663; Pu J et al. J Cell Biol. 2017. 216:4183-4197). In this latter regard, we recently showed that BORC regulates signaling by the serine/thronine kinase mTOR in response to growth factors (Jia and Bonifacino. Mol Cell. 2019. 75:26-38.e3). Binding of growth factors to cognate receptors at the cell surface initiates intracellular signaling cascades that eventually reach organelles such as lysosomes. A key component of these signaling cascades is mTOR, which exists as a subunit of two complexes named mTORC1 and mTORC2. Whereas mTORC1 associates with lysosomes, the intracellular distribution of mTORC2 is less well established. We confirmed that perinuclear clustering of lysosomes induced by uncoupling lysosomes from kinesin motors delayed the reactivation of mTORC1 by addition of serum (a source of growth factors). This finding indicated that increasing the distance of lysosome-associated mTORC1 from the source of growth factor signaling at the plasma membrane delays the relay of signals through the cytoplasm. Surprisingly, we also found that perinuclear clustering of lysosomes also delayed mTORC2 reactivation after serum replenishment. These experiments suggested the existence of pools of both mTORC1 and mTORC2 that are sensitive to lysosome positioning, a finding that may explain how changes in lysosome distribution in cancer cells promote their proliferation.
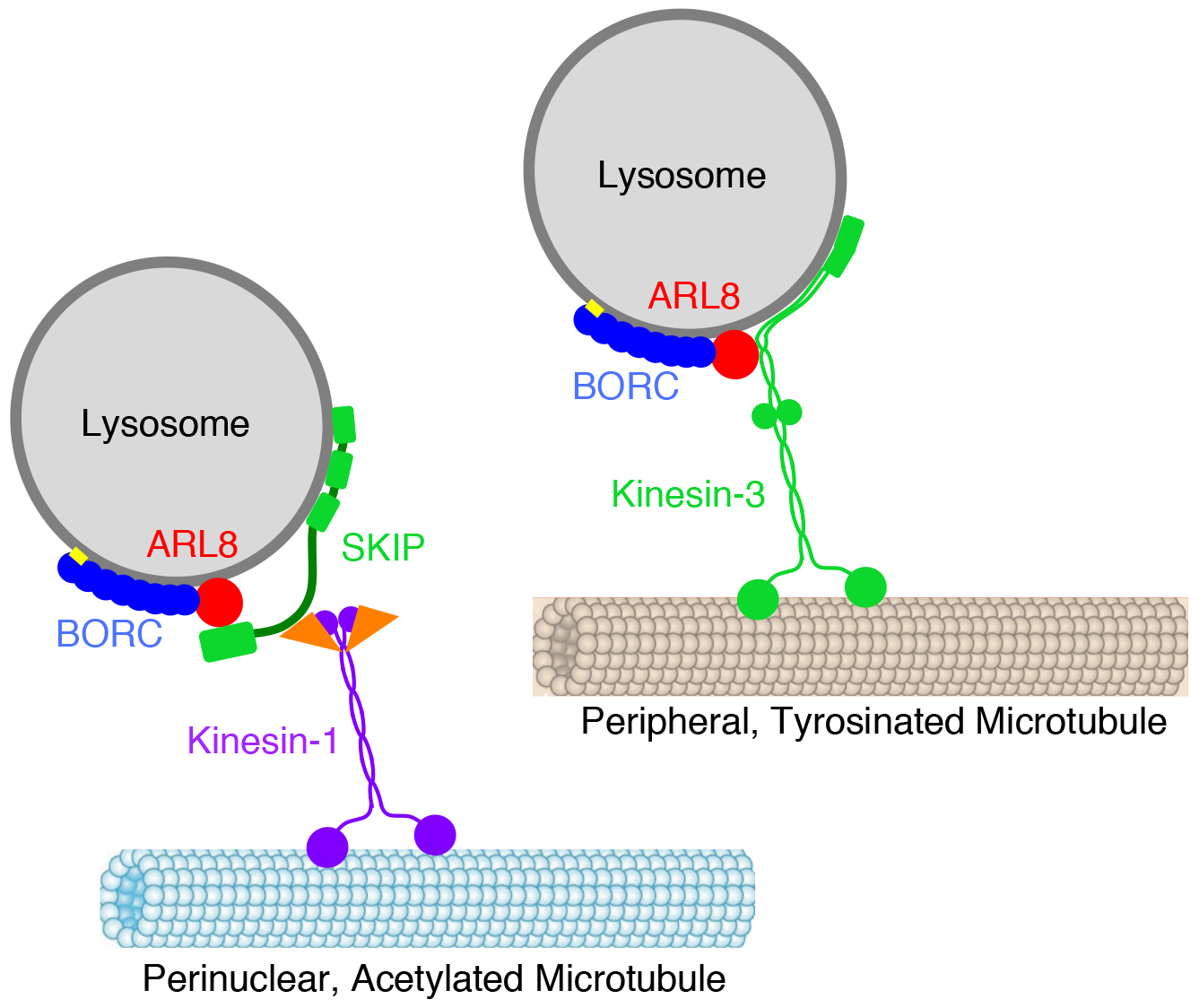
Click image to enlarge.
Reversible association with motor proteins (RAMP): a streptavidin-based method to manipulate organelle positioning
We also developed a novel method, named “reversible association with motor proteins” (RAMP), to manipulate organelle positioning within the cytoplasm (Guardia et al. PLoS Biol. 2019. 17:e3000279). RAMP consists of co-expressing (i) an organellar protein fused to the streptavidin-binding peptide (SBP), and (ii) motor domains from plus-end-directed or minus-end-directed kinesin motors fused to streptavidin (Fig. 4). The SBP-streptavidin interaction drives accumulation of organelles at the plus or minus ends of microtubules, respectively. Importantly, addition of biotin dissociates the motor from the organelle, allowing restoration of normal patterns of organelle transport and distribution. We demonstrated that this method can be used to manipulate the distribution of various intracellular organelles, including lysosomes, mitochondria, peroxisomes and the endoplasmic reticulum. This method should be useful to examine how the positioning of these organelles affects their functions, and to analyze the movement of organelle cohorts upon release from the kinesin motor.
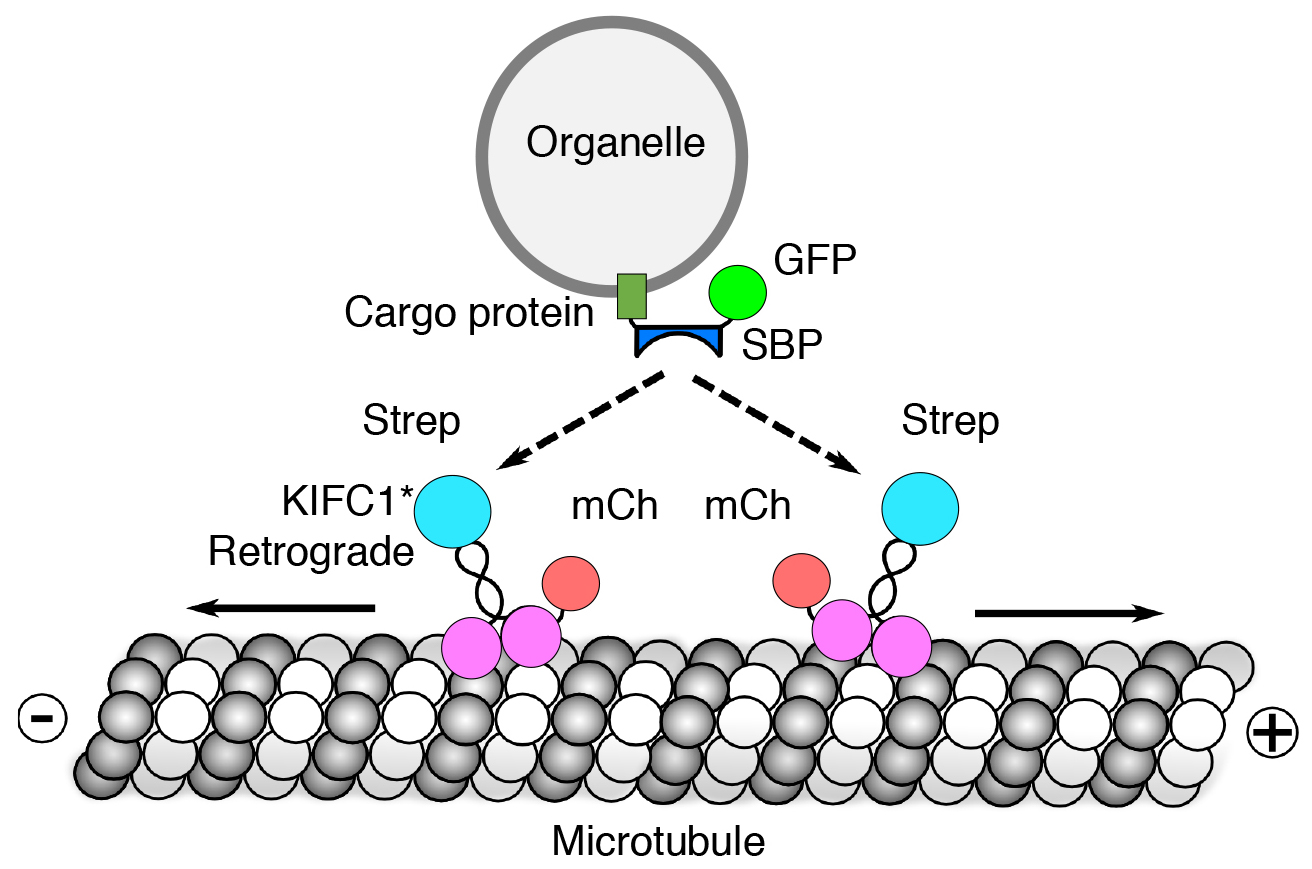
Click image to enlarge.
Mechanisms of tethering factor recruitment to the TGN
Another line of research in the laboratory deals with the mechanisms by which cargos are recycled from endosomes. This recycling involves formation of transport carriers from endosomes and their tethering and fusion to acceptor compartments such as the TGN and recycling endosomes. Tethering to these compartments is mediated by the Golgi-associated retrograde protein (GARP) and endosome-associated recycling protein (EARP) complexes, respectively. GARP is composed of VPS51, VPS52, VPS53 and VPS54 subunits, whereas EARP is composed of VPS50, VPS51, VPS52 and VPS53 subunits. While EARP is thought to be recruited to recycling endosomes by virtue of interactions with the small GTPase RAB4, up until recently the mechanism of GARP recruitment to the TGN was poorly understood. We recently found that the ARF-like (ARL) GTPase ARFRP1 is an upstream activator of two other ARL GTPases, ARL1 and ARL5, which in turn recruit golgin-type tethering factors and GARP, respectively, to the TGN (Ishida and Bonifacino. J Cell Biol. 2019. 218:3681-3696) (Fig. 5). From these findings we concluded that ARFRP1 is a master regulator of retrograde-carrier tethering to the TGN. This mechanism may be paradigmatic of other vesicular fusion events that take place within the cell.
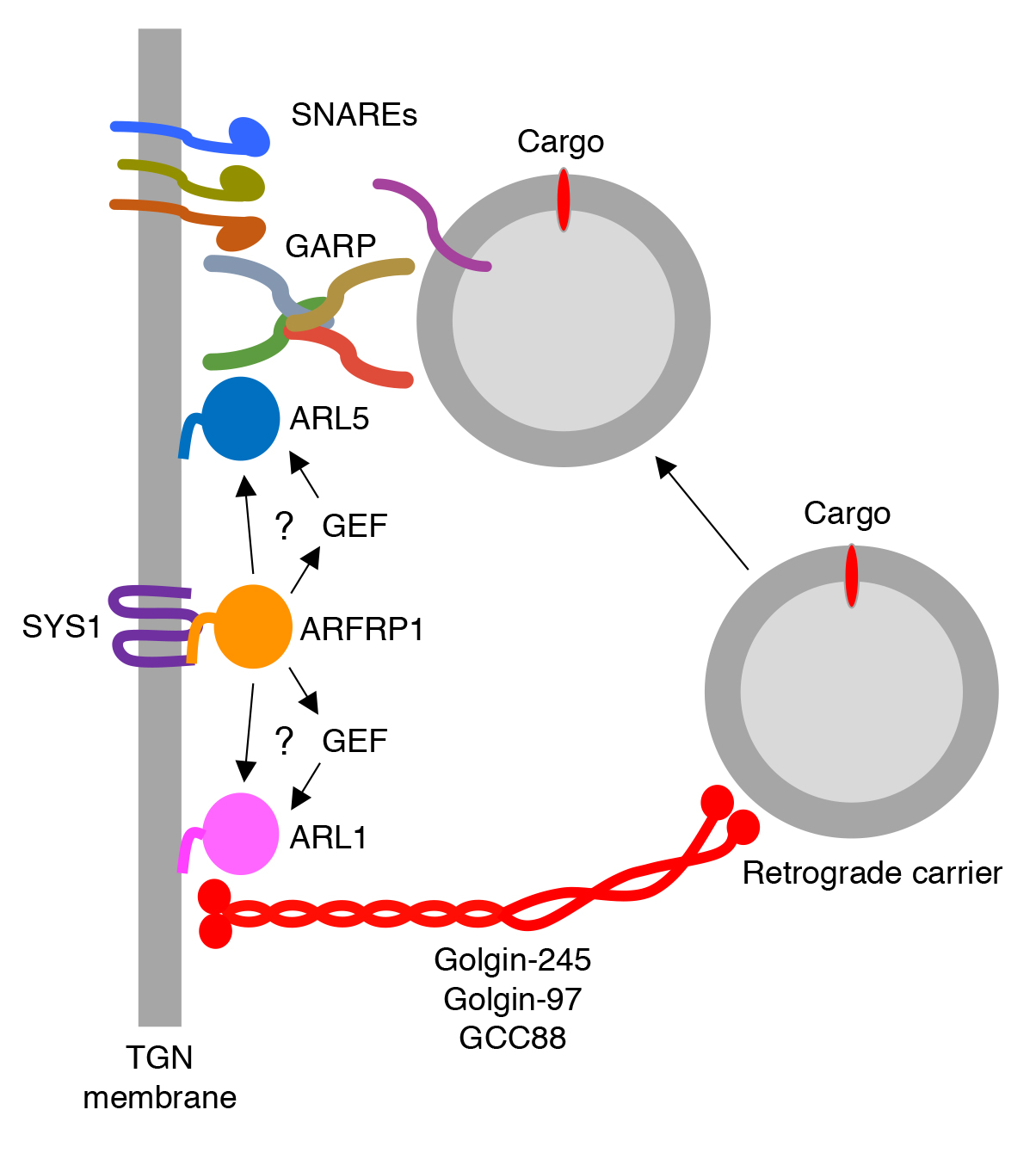
Click image to enlarge.
A novel type of pontocerebellar hypoplasia caused by mutations in the shared VPS51 subunit of the GARP and EARP complexes
Although GARP and EARP are known to play key roles in intracellular protein trafficking, their importance in human physiology remains poorly understood. In collaboration with David Everman (Greenwood Genetic Center), we recently identified compound heterozygous mutations in the gene encoding the shared GARP/EARP subunit VPS51 in a 6-year-old patient with severe global developmental delay, microcephaly, hypotonia, epilepsy, pontocerebellar abnormalities, liver dysfunction, lower extremity edema and dysmorphic features. This disease is similar to that caused by mutations in another subunit of GARP/EARP, VPS53; this disease has been variously referred to as pontocerebellar hypoplasia (PCH), progressive cerebello-cerebral atrophy (PCCA), and progressive encephalopathy with edema, hypsarrhythmia and optic atrophy (PEHO). Biochemical and cellular analyses showed that the VPS51 mutation in one allele causes a frameshift that produces a longer but highly unstable protein, whereas the mutation in the other allele produces a protein with a single amino acid substitution that is stable but assembles less efficiently with the other GARP/EARP subunits. These mutations consequently result in reduced levels of GARP and EARP complexes in the patient’s cells. In addition, the patient's cells exhibit lysosomal abnormalities, consistent with the requirement of GARP for the sorting of acid hydrolases to lysosomes. These findings thus identified a novel genetic locus for a neurodevelopmental disorder and highlighted the critical importance of GARP/EARP function in cellular and organismal physiology.
 BACK TO TOP
BACK TO TOP