Sponsored by
National Institute of Child Health and Human Development, NIH
National Institute of Neurological Disorders and Stroke, NIH
Co-sponsored by
American Academy of Pediatrics
American College of Obstetricians and Gynecologists
August 30-31, 1993
Rockville, Maryland
Edited by
Linda L. Wright, M.D.
Pregnancy and Perinatology Branch
Center for Research for Mothers and Children
National Institute of Child Health and Human Development, NIH
Gerald B. Merenstein, M.D.
University of Colorado
Health Sciences Center
The Children's Hospital
Deborah Hirtz, M.D.
Developmental Neurology Branch
National Institute of Neurological Disorders and Stroke, NIH
U.S. Department of Health and Human Services
Public Health Service
National Institutes of Health
National Institute of Child Health and Human Development
NIH Publication No. 96-3823
March 1996
Contents
Executive Summary
Gerald B. Merenstein
Participants
Session I: Scientific Basis of Brain Injury in Acute Perinatal Asphyxia
Moderator: Gerald B. Merenstein
Mechanisms By Which Asphyxial Insults Destroy Neurons
Peter D. Gluckman, Christopher E. Williams,
William K.M. Tan, E. Carina Mallard
Fetal Brain Metabolism Under Stress—Oxygenation, Acid-Base and Glucose
Julian T. Parer
Cellular Alterations Associated With Perinatal Asphyxia
Michael V. Johnston
Hypoxia and Cerebral Blood Flow in the Human
Gorm Greisen
Hypoxia Opportunism During Brain Development
Philippe Evrard, Jean-François Gadisseux, Pierre Gressens
Discussants
Ingemar Kjellmer
Philippe Evrard
Session II: Clinical Assessment— Obstetrics
Moderator: Edward J. Quilligan
Fetal Monitoring: Utility and Interpretation of Umbilical Cord Blood Gases and Fetal Scalp Sampling
John C. Hauth
Fetal Monitoring: Role of Doppler Blood Flow Velocity and Fetal Heart Rate in Assessment of Fetal Asphyxia
James A. Low
Perinatal Asphyxia and Placental Pathology
Carolyn M. Salafia
Discussants
Nigel Paneth
Julian T. Parer
Session III: Clinical Assessment-— Neonatal
Moderator: Linda L. Wright
Neuroimaging of Perinatal Asphyxia in Term Infants
Marvin D. Nelson, Jr.
Differential Diagnosis and Contribution of EEG
Robert R. Clancy
Neonatal Diagnosis of Perinatal Asphyxia
Gerald B. Merenstein, Brian S. Carter
Discussants
John Freeman
William Oh
Session IV: Interventions
Moderator: Deborah Hirtz
Acute Perinatal Asphyxia—Conventional Management
James A. Lemons
Neuronal Rescue and Neuronal Prophylaxis—Studies in Animals
Peter D. Gluckman, Christopher E. Williams, Barbara M. Johnston, Jian Guan,
Ernest S. Sirimanne, William K.M. Tan
Glutamate Antagonists, Calcium Channel Blockers and Allopurinol
Malcolm I. Levene
Discussants
William W. Hay, Jr.
Michael V. Johnston
Session V: Clinical Studies of Long- Term Outcome
Moderator: John Freeman
Long-Term Outcome in Birth Asphyxia—The Role of Neonatal Encephalopathy in Prediction
Karin Nelson
Assessment of Outcomes Following Neonatal Asphyxia
Jane E. Stewart, Marie C. McCormick, Alan Leviton
Discussants
Alan Leviton
James A. Lemons
Session VI: Clinical Research
Moderator: Jerold F. Lucey
Methodologies for Documenting Timing and Evidence of
Brain Asphyxia Including NIR and NMR
A.D. Edwards
Clinical Research Methodology for Studies Based on
Consensus Definition of Birth Asphyxia
John C. Sinclair
Discussants
William Oh
Jerold F. Lucey
Executive Summary
Gerald B. Merenstein, M.D.
Acute Perinatal Asphyxia in Term Infants
An international workshop on Acute Perinatal Asphyxia in Term Infants was convened by the NICHD and the NINDS and cosponsored by the AAP and ACOG in Rockville, MD on August 30-31, 1993. The purpose was to review the current knowledge of the definition and diagnosis of acute perinatal asphyxia in term infants in order to develop operational and specific criteria to be tested in new studies of acute perinatal asphyxia. The workshop summarized information currently available on this topic and specifically addressed the following questions: Is there a consensus definition of acute perinatal asphyxia in the term infant? If not, what research is needed to develop one? What research is needed to validate a research definition? What research is needed to assess the relationship of acute perinatal asphyxia in the term infant to both short- and long- term outcome?
Session I: Scientific Basis of Brain Injury in Acute Perinatal Asphyxia
Peter Gluckman first addressed mechanisms by which metabolic insults destroy neurons. He emphasized that there are a variety of mechanisms, some of which are operative during the insult (primary neuronal death), others immediately after injury (reactive cell death or reperfusion injury), and others hours or days later (delayed neuronal death). Prophylactic treatment must focus on interference with mechanisms operative during or immediately after insult, while neuronal rescue needs to focus on mechanisms before or during the initial stages of delayed neuronal death.
Certain factors alter the sensitivity of the brain to injury. These include gestational age, intrauterine growth restriction, metabolic factors, and brain temperature. The role of glucose is controversial; its effects may depend on the nature of the insult and overall metabolic status. Acidosis may be due to an increase in lactate or H+ concentration. Failure of pH buffering may lead to depolarization of neurons, energy failure, and loss of calcium and sodium homeostasis. Moderate acidosis may beneficially suppress activity of excitatory NMDA receptors. Temperature elevations are associated with increased injury and hypothermia is associated with decreased injury. Single insults and repeated short insults cause injuries to different parts of the brain.
Clinical trials of interventions must address the phase of the injury. Therapy for reperfusion injury must be given before the insult or early during the insult. Neuronal rescue strategies to arrest apoptosis (programmed cell death), to reduce the inflammatory response, and to suppress post-asphyxial seizures seem most promising.
Julian Parer discussed fetal brain metabolism under stress, including oxygen consumption, acidosis, and energy metabolism. He noted that oxygen consumption is constant over a wide range of oxygen content in arterial blood due to changes in cerebral blood flow (CBF) which matches changes in oxygen content and extraction. Experimental results indicate that in the sheep model oxygen delivery is 70 percent greater than that in the adult; thus, fetal CBF is higher than adult CBF at any arterial oxygen content.
Michael Johnston addressed cellular alterations with perinatal asphyxia. Asphyxia triggers a cascade of cellular biochemical events which lead to temporary alterations in cellular function and/or cell death. Hypoxia leads to a depolarization of neuronal membranes, alterations in cellular ion homeostasis, and changes in energy metabolism. There is an increased release and diminished reuptake of neurotransmitters including the excitatory amino acid glutamate and an abnormal accumulation of intracellular calcium which kills cells by activation of proteases, lipases, protein kinase C and generation of free radicals.
Implications for management include support of CBF, maintenance of plasma glucose concentrations and suppression of seizures. Further therapy should be based on an understanding of the cascade of events. Future therapies may include glutamate antagonists, calcium channel antagonists, and drugs that limit intracellular calcium release, singly or in combination, based on the mechanism and timing of the pathologic events.
Gorm Griesen presented information on hypoxia and CBF in humans. Resting CBF is low in the human neonate (30-50 percent) compared to adult values. Data on the acute effects of hypoxia on CBF in the human fetus and newborn is extremely limited. In acute hypoxemia, the combination of increases in CBF by two- to threefold and increased oxygen extraction allows electrical function to persist until arterial oxygen saturation falls below 50 percent. Asphyxia results in the redistribution of CBF, mediated by the sympathetic system, in the perinatal animal brain. Preliminary evidence indicates that this may not necessarily be so in the human. In human infants delayed luxury perfusion with lost cerebrovascular reactivity is associated with a grave prognosis. It is not clear if this represents a cause or result of neuronal damage.
Phillipe Evrard discussed the influence of hypoxia on developing neural tissue. He emphasized the need for research on the precise time of the insult, the chronology of regional angiogenesis, and difference in metabolism of different populations of neuronal cells, as well as the diverse pathogenic mechanisms affecting cytoarchitechtonic integrity necessary for normal neurodevelopment.
Session II: Clinical Assessment—Obstetrics
John Hauth addressed the utility and interpretation of umbilical cord blood gases and fetal scalp sampling in fetal monitoring. Umbilical cord blood acid base assessment is an objective measure of the status of the newborn. In the absence of newborn metabolic acidemia it is a physiologic certainty that proximate fetal hypoxia did not occur. Although normal ranges for umbilical artery pH and blood gas values have been established, the degree of severity of newborn metabolic acidemia associated with neonatal seizures, prolonged hypotonia, or multi-organ system dysfunction is not known.
The duration and extent of hypoxia that will result in metabolic acidosis and neurologic damage to the fetus are not known, nor have the lower limits of umbilical artery pH (metabolic acidemia) and degree of depression (low 5-minute Apgars) that are predictive of subsequent neurologic dysfunction been determined. Increased neonatal morbidity is related to asphyxia in term newborns when asphyxia is defined by umbilical cord pH < 7.0 (including metabolic acidemia) and 5-minute Apgar score of £ 3. Umbilical cord blood gas sampling is used much more frequently than fetal scalp sampling in the United States today.
James Low reviewed the role of Doppler blood flow velocity and fetal heart rate (FHR) measurements in fetal monitoring. In selected pregnancies Doppler blood flow velocity2 and FHR are predictive of fetal asphyxia but have limited predictive value for asphyxia- induced brain damage. They do not have a role as a screening test for clinical fetal distress in the general obstetric population. In growth retarded infants, abnormal umbilical artery blood flow velocity is associated with hypoxemia, hypercapnia, and metabolic acidosis. Abnormal nonstress tests and low biophysical profiles are associated with abnormalities of reproductive outcome but randomized clinical trials have failed to demonstrate improved outcome with intermittent antepartum non-stress tests in high-risk pregnancies. Fetal cardiovascular response to hypoxemia includes reduced heart rate variability and heart rate deceleration as well as decreased fetal body movements and fetal breathing. Major problems of reliability of interpretation of visually-read intrapartum recordings make their use questionable; computerized intrapartum FHR reading may standardize interpretation of FHR patterns. Because the prevalence of fetal asphyxia and brain damage due to acute asphyxial insults is low, large randomized clinical trials will be required to document that assessment measures will change the incidence of severe asphyxial insults associated with adverse neurologic outcome.
Carolyn Salafia addressed the placental pathology associated with perinatal asphyxia. She reviewed placental anatomy, physiology, and biochemistry, emphasizing the complexity of the placenta, the multiple pathophysiologies associated with acute or chronic perinatal asphyxia, the lack of specificity of indicators of placental damage, and the imprecision of histologic timing of antenatal events. The placenta may provide a background to assist interpretation of the sequence of events leading to acute perinatal asphyxia.
Session III: Clinical Assessment—Neonatal
Marvin Nelson noted that the high water content and incomplete myelination of the term human brain make discrimination between white and gray matter difficult on neuroimaging. The contours of the brain surface and the lateral ventricles are well visualized. Serial ultrasounds are recommended to evaluate the acutely damaged neonatal brain.
Robert Clancy explored issues pertinent to the differential diagnosis of neonatal encephalopathy and contribution of the EEG. Neonatal encephalopathy may arise from acute disorders that generally provoke acute EEG changes. Chronic disorders such as cerebral dysgenesis are associated with long-standing or chronic EEG changes. Peripheral disorders such as Werdnig-Hoffman disease may mimic an encephalopathy but preserve mental status and have a normal EEG.
The EEG is a sensitive, but non-specific measure of whole brain function; EEG abnormalities are not specific for etiology of neonatal encephalopathy or reversibility. EEG background abnormalities at the height of an encephalopathy have increased prognostic power; changes that persist and endure beyond the insult are more clinically significant of severe abnonnalities associated with poor prognosis.
Gerald Merenstein reviewed the neonatal diagnosis of perinatal asphyxia. CNS injury in the newborn has multiple etiologies including intrapartum hypoxia, intracranial hemorrhage (ICH), metabolic disorders, drug withdrawal, congenital viral infection, acute viral and bacterial infection, neurodevelopmental defects, and others.
Signs of hypoxic-ischemic encephalopathy (HIE) include seizures; apnea; respiratory arrest; hyperalertness; jitteriness; posturing, movement disorders; impaired suck, swallow, gag and feeding; hypotonia; and abnormal oculomotor and pupillary response. The proportion of HIE due to ischemia is unknown.
The need for a usable definition of perinatal asphyxia was emphasized by a review of the abstracts on perinatal asphyxia presented at the 1993 APS/SPR meetings where nine different definitions were used for various studies. The AAP/ACOG definition is useful but excludes infants with perinatal asphyxia without neurologic involvement. Its usefulness is also limited since it requires observation of the newbom for varying periods before a diagnosis can be made. An asphyxial scoring system using electronic fetal monitoring, cord gases, and 5-minute Apgar score yielded excellent specificity and positive predictive value for dysfunction of three or more organ systems.
Session IV: Interventions
Conventional management of acute perinatal asphyxia was reviewed by James Lemons. Although there is a lack of appropriate evaluation through randomized trials, conventional management currently includes prompt and expert resuscitation, support of the cardiovascular and respiratory systems, careful monitoring of basic metabolic and hematologic parameters, vigorous treatment of seizures, attention to other organ system injury, avoidance of unproved therapy and timely intervention with conventional management including circulatory and metabolic support. Newer therapies warrant careful assessment as the mechanisms underlying the cellular and molecular pathophysiology of asphyxia become clearly defined. Prevention is the most effective but most elusive strategy to decrease perinatal morbidity secondary to birth asphyxia.
Peter Gluckman addressed neuronal rescue and neuronal prophylaxis studies in animals. Primary considerations for rescue or prophylaxis include the temporal relationship of the intervention to the nature and timing of the putative insult, i.e., IGF-l is effective if given after but not before an injury, and the influence of certain factors, i.e., calcium channel blockers may be neuroprotective but the secondary effects on the cardiovascular system may lead to hypotension and aggravation of the insult. Failure to consider the mechanism of action of the therapy, the relative timing of the therapy and insult, and the potential impact of side effects of the therapy confound the results of animal research and make extrapolation to potential clinical settings difficult.
Malcolm Levene reviewed the calcium transport systems and glutamate stimulation of the quisqualate/kainate and NMDA receptors. Asphyxia causes excessive calcium entry into the neuron, primarily related to glutamate over-release, a cascade of enzymes, and cell-death. The major approaches to neuroprotection have been preventing calcium entry.
Experimental treatments in humans include glutamate antagonists, calcium channel blockers, and allopurinol. Neuronal protection following birth asphyxia by means of pharmacological agents remains controversial.
Session V: Clinical Studies of Long-term Outcome
Karin Nelson explored long-term outcome in birth asphyxia and the role of neonatal encephalopathy in prediction. The major predictor of long- term neurologic outcome is neonatal encephalopathy. There is a need for more information on the differential diagnosis of neonatal encephalopathy and the natural history of its specific sub-syndromes. More information is needed on differential diagnosis and the natural history of combinations of factors proposed for definition in birth asphyxia (e.g., Apgar score and acidosis). Efforts must be made to increase the specificity of identifiable risk factors since irreversible brain damage associated with acute perinatal asphyxia in term infants is very uncommon.
Marie McCormick addressed the topic of asphyxia as it pertains to long-term outcome. She noted that studies of outcomes of infants with perinatal asphyxia remain cumbersome due to differences in the basic definition of asphyxia, differences in measuring outcome, and differences in evaluating confounding factors. Accurate classification of the severity of neonatal encephalopathy improves the ability to predict long-term outcome.
Session VI: Clinical Research
David Edwards presented information on methodologies for documenting timing and evidence of brain asphyxia including magnetic resonance spectroscopy (NMR), near infrared spectroscopy (NIR), and cerebral electrophysiological measurements. Phosphorus NMR spectra in infants with HI brain injury regularly show impaired oxidative phosphorylation as measured by the phosphocreatine to inorganic phosphate ratio (PCr/Pi) as well as an ATP concentration below normal. The spectrum may be normal immediately after resuscitation; abnormalities gradually reach a maximum at about 3 days. Recovery of the spectrum takes place over approximately 2 weeks in surviving infants. In the newborn pig model subjected to HI, there is a rapid fall in PCr/Pi, pH, and eventually ATP One or 2 hours after resuscitation the alterations return to near baseline values; abnormal PCr/Pi similar to infants with HIE develop some 8 hours later.
John Sinclair discussed clinical research methodology for studies based on consensus definition of birth asphyxia, what constitutes a valid, reliable and clinically-applicable definition of birth asphyxia, what questions would we ask, and what are the methodological requirements designed to answer these questions.
There may be two forms of a clinically useful standard definition—one for studies under highly controlled circumstances and one for field or population-based studies. The first would include state-of-the-art technology that is sophisticated, invasive and expensive and only available in certain centers and for use only in highly selected patients. The second would include routinely available, non-invasive, inexpensive technology which may be applied to large populations.
Four types of research are addressed: 1) evaluation of putative causes of birth asphyxia, 2) evaluation of diagnostic tests for birth asphyxia, 3) assessment of natural history of birth asphyxia, and 4) evaluation of treatment and prevention of birth asphyxia.
Toward a Research Definition of Perinatal Asphyx
The final afternoon of the workshop was devoted to open discussion by all workshop attendees. Although consensus was not reached on a definition of acute perinatal asphyxia, there were several areas where the majority of participants agreed.
The majority of participants felt the need for further animal research to better delineate pathophysiology of acute perinatal asphyxia and for investigation of potential interventions. It is probable that a consistent insult does not invariably result in a single pathological response. It will be important to establish the integrity of the placental-fetal unit before labor and delivery, to identify fetuses which are vulnerable to ischemic insult, and to differentiate chronic underlying abnormalities from acute events. There is also a need to define and implement a rapid, aggressive workup for non-asphyxial etiologies in the encephalopathic newborn. Finally, timing and pattern of the insult and the use of interventions must be interrelated.
Most participants felt that human therapeutic drug trials were premature at this time. When drug trials from animals are extrapolated to clinical trials in humans the following issues must be considered: risk of side effects, risk of toxic interactions, understanding of temporal relationship of insult and interventions, response at different maturational levels, and appropriate outcome measures. It will be important to evaluate a spectrum of cognitive and motor outcomes in children at least 18 months old using standardized measures. The clinical research methodologies presented by Dr. Sinclair, including randomization, intention-to-treat analysis, long-term follow-up, and double masking (to the intervention and outcome) were generally agreed to be important. In addition, results should be reported as point estimates and 95% confidence intervals using both relative and absolute estimators of treatment effect, i.e., relative risk, relative risk reduction, absolute risk reduction, and the inverse of absolute risk reduction or number needed to treat in order to prevent one adverse target outcome.
Use of the AAP/ACOG definition is limited by the time required for development of organ-specific signs, but following severely affected infants so defined may, nevertheless, be of value. The use of the combined pH <7.0 and 5-minute Apgar of £ 3 or the use of the scoring system utilizing FHR monitoring, cord acid-base status, and 5-minute Apgar score might be useful in studies of long-term morbidities, including tests of sensitivity and specificity. Performance of diagnostic modalities which provide physiologic correlates of brain function such as early EEG, NIRS, NMR before secondary injury (at about 8 hours) in an identified high-risk group may provide the earliest markers for secondary events and the predictive accuracy of diagnostic tests for long-term outcome may be proven. A diagnostic modality which accurately indicates that inadequate oxygenation is about to occur or is occurring would be very useful. Current diagnostic modalities define variable physiology, some of which may be true pathological physiology, but they do not define what variations and what pathologies cause injury. Other definitions of perinatal asphyxia should not be utilized unless they are first validated.
Participants
Acute Perinatal Asphyxia Conference
August 30-31, 1993, Rockville, Maryland
Scott Andres, Ph.D.
Pregnancy and Perinatology Branch
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B03C
Bethesda, Maryland 20892
FAX: 301-496-3790
TEL: 301-496-5575
F.J. Brinley, Jr., M.D., Ph.D.
Division of Convulsive, Developmental,
and Neuromuscular Disorders
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 816
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402-0302
TEL: 301-496-6541
Sarah H. Broman, Ph.D.
Division of Convulsive, Developmental,
and Neuromuscular Disorders
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 8C06
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402-0887
TEL: 301-496-5821
Brian Carter, M.D.
Department of Pediatrics
Fitzsimons Army Medical Center
Aurora, Colorado 80045-5001
FAX: 303-361- 4278
TEL: 303-361-8192
Charlotte Catz, M.D.
Pregnancy and Perinatology Branch
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B03E
Bethesda, Maryland 20892
FAX: 301-496-3790
TEL: 301-496- 5575
Robert R. Clancy, M.D.
Department of Neurology
Children's Hospital of Philadelphia
34th Street & Civic Center Boulevard
Philadelphia, Pennsylvania 19104-4399
FAX: 215-590-1771
TEL: 215-590-1719
Sue Davis, M.D.
Department of Pediatrics
School of Medicine
University of Auckland
Private Bag 92 019
Auckland, New Zealand
FAX: 011-64-9-373-7481
TEL: 011-64-9-373-7999 x6450
Joseph S. Drage, M.D.
Division of Convulsive, Developmental,
and Neuromuscular Disorders
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 816
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402-0302
TEL: 301-496-6541
A.D. Edwards, M.D.
Department of Paediatrics and Neonatal Medicine
Royal PostGraduate Medical Centre
Hammersmith Hospital
Du Cane Road
London, England W12 ONN
FAX: 44-081-740-8281
TEL: 44-081-740-3326
Philippe Evrard, M.D.
Universite Catholique de Louvain
Clinques Universitaires Saint-Luc
Avenue Hippocrate l0UCI 10/1303
1200 Bruxelles
Belgium
FAX: 011-32-2-764-52-31
TEL: 011-32-2-764-10-62/68
John Freeman, M.D.
Johns Hopkins Hospital
CMSC 1-141
600 North Wolfe Street
Baltimore, Maryland 21287-3141
FAX: 410-614-0373
TEL: 410-955-9100
Peter D. Gluckman, M.D.
Dean's Office
School of Medicine
University of Auckland
Private Bag
Auckland, New Zealand
FAX: 011-64-9-3737-482
TEL: 011-64-9-3737-521
Gorm Greisen, M.D.
Department of Neonatology
Rigshospitalet
Blegdamsvej 9
2100 Copenhagen O
Denmark
FAX: 45- 3545-5025
TEL: 45-3545-3545
John C. Hauth, M.D.
Department of OB/GYN
University of Alabama at Birmingham
University Station
Birmingham, Alabama 35233-7333
FAX: 205-975-4375
TEL: 205-934-5611
William W. Hay, Jr., M.D.
University of Colorado Health Sciences Center
4200 East 9th Avenue, Box B-195
Denver, Colorado 80262
FAX: 303-270-8067
TEL: 303-270-5981
Deborah Hirtz, M.D.
Developmental Neurology Branch
National Institute of Neurological
Disorders and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 8C02
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402- 0887
TEL: 301-496-5821
Michael V. Johnston, M.D.
Johns Hopkins University
School of Medicine
Kennedy Krieger Institute
707 North Broadway
Baltimore, Maryland 21205
FAX: 410-550-9524
TEL: 410-550-9492
Ingemar Kjellmer, M.D., Ph.D.
Gothenburg University
Department of Paediatrics
East Hospital
S-416 85 Goteburg
Sweden
FAX: 46-031-84-3010
TEL: 46-031-37-4631
Tracy Lawrence-Black, M.D.
McMaster University Medical Centre
Department of Pediatrics
Room 4G40C
1200 Main Street West
Hamilton, Ontario, Canada L8N 3Z5
FAX: 416-521-5007
TEL: 416-521-2100 x5611
James A. Lemons, M.D.
Section on Neonatal-Perinatal Medicine
Indiana University Medical Center
702 Barnhill Drive
Indianapolis, Indiana 46202-5210
FAX: 317-274-2065
TEL: 317-274-4716
Malcolm I. Levene, M.D.
D Floor, Clarendon Wing
The General Infirmary at Leeds
Belmont Grove, Leeds LS29NS
England
FAX: 44 0532 316021
TEL: 44 0532 432799 x3905
Alan Leviton, M.D.
Children's Hospital
300 Longwood Avenue
Boston, Massachusetts 02115
FAX: 617-735-7429 [attn: x6492]
TEL: 617-735-6491
James A. Low, M.D.
Kingston General Hospital
76 Stuart Street
Watkins II
Kingston, Ontario K7L 2V7
Canada
FAX: 613-548-1330
TEL: 613-548-1381
Jerold F. Lucey, M.D., F.A.A.P
Department of Pediatrics
Medical Center Hospital of Vermont
McClure 718
111 Colchester Avenue
Burlington, Vermont 05401
FAX: 802-656-4844
TEL: 802-862-8778
Marie C. McCormick, M.D., Sc.D.
Department of Maternal and Child Health
Harvard School of Public Health
677 Huntington Avenue
Boston, Massachusetts 02115
FAX: 617-432-3755
TEL: 617-432-1080
TEL: 617-735-8330 Thursday only
Donald McNellis, M.D.
Pregnancy and Perinatology Branch
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B03H
Bethesda, Maryland 20892
FAX: 301-496-3790
TEL: 301-496-5575
Gerald B. Merenstein, M.D.
University of Colorado
Health Sciences Center
The Children's Hospital
Campus Box B065
1056 East Nineteenth Avenue
Denver, Colorado 80218
FAX: 303-837-2729
TEL: 303-837-2703
Eli M. Mizrahi, M.D.
The Methodist Hospital
Baylor College of Medicine
6565 Fannin Street
Houston, Texas 77030
FAX: 713-793-1574
TEL: 713-790-3105
Karin Nelson, M.D.
Neuroepidemiology Branch (NEB)
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 714
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-496- 2358
TEL: 301-496-1714
Marvin D. Nelson, Jr., M.D.
Department of Radiology
Children's Hospital of Los Angeles
4650 Sunset Boulevard #81
Los Angeles, California 90027
FAX: 213-666-7816
TEL: 213-669-4572
William Oh, M.D.
Women & Infants Hospital of Rhode Island
101 Dudley Street
Providence, Rhode Island 02905
FAX: 401-453-7571
TEL: 401-274-5983
Nigel Paneth, M.D., M.P.H.
Michigan State University
A206 East Fee Hall
East Lansing, Michigan 48824-1316
FAX: 517- 336-1130
TEL: 517-353-8623
Julian T. Parer, M.D., Ph.D.
Department of OB/GYN
University of California, San Francisco
513 Parnassus, Room HSE 1462
Box 0550
San Francisco, California 94143
FAX: 415-476-9266
TEL: 415-476-2945
Edward J. Quilligan, M.D.
Department of OB/GYN
UC Irvine, Medical Center
101 The City Drive, South
Building 26, Route 81
Orange, California 92668
FAX: 714-456-6073
TEL: 714-456-6823
Dwight Rouse, M.D.
Department of OB/GYN
University of Alabama at Birmingham
University Station
Birmingham, Alabama 35233-7333
FAX: 205-975-4375
TEL: 205-934-5611
Carolyn M. Salafia, M.D.
Perinatology Research Branch
National Institute of Child Health
and Human Development (NICHD)
Bethesda, Maryland 20892
FAX: 202-784-1382
TEL: 202-784-0755
Philip H. Sheridan, M.D.
Developmental Neurology Branch
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 8C10
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402- 0887
TEL: 301-496-6701
John C. Sinclair, M.D.
Department of Pediatrics
McMaster University Medical Centre
Room 4G40C
1200 Main Street West
Hamilton, Ontario, Canada L8N 3Z5
FAX: 416-521-5007
TEL: 416-521-2100 x5611
Giovanna M. Spinella, M.D.
Developmental Neurology Branch
National Institute of Neurological Disorders
and Stroke (NINDS)
National Institutes of Health
Federal Building, Room 820
7550 Wisconsin Avenue
Bethesda, Maryland 20892
FAX: 301-402- 0887
TEL: 301-496-5821
Jane E. Stewart, M.D., M.S.
Children's Hospital
Joint Program in Neonatology
300 Longwood Avenue
Boston, Massachusetts 02115
FAX: 617-732-4151
TEL: 617-732-4180
Marian Willinger, Ph.D.
Pregnancy and Perinatology Branch
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B03D
Bethesda, Maryland 20892
FAX: 301-496- 3790
TEL: 301-496-5575
Linda L. Wright, M.D.
Pregnancy and Perinatology Branch
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B03F
Bethesda, Maryland 20892
FAX: 301-496-3790
TEL: 301-496-5575
Sumner J. Yaffe, M.D.
Center for Research for Mothers
and Children (CRMC)
National Institute of Child Health and
Human Development (NICHD)
National Institutes of Health
6100 Executive Boulevard, Room 4B05K
Bethesda, Maryland 20892
FAX: 301-402- 2085
TEL: 301-496-5097
Session I: Scientific Basis of Brain Injury in Acute Perinatal Asphyxia
Moderator: Gerald B. Merenstein
Mechanisms By Which Asphyxial Insults Destroy Neurons
Peter D. Gluckman, M.D., Christopher E. Williams, Ph.D., William K.M. Tan, Ph.D., and E. Carina Mallard, B.Sc.
Research Centre for Developmental Medicine and Biology, University of Auckland, New Zealand
Background
Experimental evidence shows that when neurons suffer an hypoxic-ischemic (HI) injury, a variety of mechanisms play a role in neuronal loss.1 Some are operative during the insult itself and lead to immediate neuronal loss (primary neuronal death), but if the insult is reversible, then other mechanisms are operative either immediately after the injury (reactive cell death or reperfusion injury) or hours or days later (delayed neuronal death).2, 3
The relative importance of the mechanisms operative at each of these phases clearly depends on the nature, the degree, the length and reversibility of the insult. It is these differences, along with the associated differences in metabolic milieu, that are likely to underlie the well recognized differences in therapeutic terms between experimental global and focal asphyxia.
From the perspective of selecting potential therapeutic interventions, an understanding of the temporal or pathophysiologic phases of injury is essential. Clearly prophylactic therapy should focus on interference with mechanisms operative during or immediately after the insult. Neuronal rescue therapies should focus on mechanisms operative during delayed neuronal death. For example insulin-like growth factor-1 (IGF-1), which is presumed to interfere with apoptosis, is active if administered after a HI insult4 but not if given before.5 Conversely flunarizine, a calcium channel blocker, is active if given before6 but not after a similar injury.7 Too few experimental studies have recognized the importance of this temporal consideration which makes interpretation of experiments where, for example, the agent is started before and continued after the insult difficult to interpret mechanistically.
Sensitizing Factors
A number of factors clearly alter the sensitivity of the brain to injury—they not only confound interpretation of experiments but also must be considered in defining intervention stratagems.
Gestational Age
The maturational stage of the developing brain may be important. There is evidence that, for a standard insult, the extremely immature brain may be more resistant than the mature brain. For example at 90 days gestation (term 147 days) in the fetal sheep, 10 minutes of total umbilical cord occlusion causes no injury, whereas at 120 days after most neurogenesis is complete and myelinization has started, significant hippocampal loss is seen.8,9 Presumably the increased resistance of the gray matter to injury results from the lower metabolic rates and enhanced ability of the immature cerebrum to maintain K+ gradients.10 This change in sensitivity may also depend, in part, on the greater depressive effect on the cardiovascular system at the older age.9
Intrauterine Growth Retardation (IUGR)
Neural maturation is altered in IUGR with evidence of dysmaturation with some aspects delayed and some apparently precocial.11,12 Limited evidence suggests a greater sensitivity to asphyxial injury13—in part this is probably due to the associated alterations in metabolic milieu.
Metabolic Factors
The role of glucose and the developing brain has been controversial—whereas in the adult, hyperglycemia clearly aggravates the injury, in the immature Levine rat model there may be a degree on protection by hyperglycemia.14,15 On the other hand glucose administration can be toxic, for example, to the growth retarded fetus and lactic acidosis may aggravate the injury (unpublished observations). Thus the effects of glucose administration may depend on the nature of the insult and overall metabolic status. The consequences of lactic acidosis may be due to magnitude of changes in H+ rather than the rise in lactate. Failure of pH buffering under very severe conditions is closely coupled with depolarization of neurones, energy failure and loss of calcium and sodium homeostasis.16 Alternatively moderate acidosis may suppress activity of the excitatory NMDA receptor and protect developing neurons against hypoxia in culture.17,18
Brain Temperature
There is clear experimental evidence that small increases in temperature are associated with more severe injury and moderate hypothermia of, for example, 2C° can provide relative protection.19 Indeed hypothermia is the mode of action of some neuroprotective agents. Presumably hypothermia acts by reducing metabolic demands and perhaps by altering cerebral/peripheral blood flow demands. Given the hyperthermia of the in utero environment, this is a further argument for delivery of the compromised fetus and offers a simple potential therapeutic approach because the degree of protection conferred by mild hypothermia is quite marked in the adult and developing brain.
The Pattern of Injury
Whether the injury is focal (e.g., stroke like) or global (e.g., systemic asphyxia and hypotension) has obvious effects on the distribution of neuronal loss. Primary mechanisms of injury dominate in the core of a focal injury whereas the surrounding penumbra tends to respond differently to therapeutic intervention. More recently we have shown that single insults in late gestation fetal sheep produce hippocampal and/or cortical loss,8,20,21 whereas repeated short insults cause a shift to striatal injury.22 The basis of this difference is not yet known but clearly may have important mechanistic and therapeutic implications.
Mechanisms Leading to Neuronal Death
A number of mechanisms have been implicated in neuronal death secondary to HI injury. When considering the mechanisms of damage it is important to remember that two distinct patterns of cell loss can be seen after HI injury, namely selective neuronal loss and infarction. Selective neuronal loss is apparent as loss of susceptible neurons individually and shares many characteristics with apoptosis, namely chromatin fragmentation, condensation and cytoplasmic shrinkage. In contrast both glia and neurons are lost during infarction and the lesion is apparent as an area of total tissue necrosis.
Energy Failure
Impaired substrate delivery can lead to energy failure and acute injury. This energy failure is initially reversible if substrate delivery is rapidly restored. Some hours after a severe injury there can be a secondary loss of high energy metabolites.23 Presumably this energy failure corresponds to the mitochondrial disruption that occurs with the development of infarcts. The precise cause of the secondary deterioration is unclear although it is unlikely to be due to impaired delivery of glucose since extracellular concentrations of glucose rise after a severe HI injury in the developing brain (unpublished result).
Intracellular Edema and Membrane Damage
Intracellular ion homeostasis is energy dependent. Energy failure and depolarization together lead to intracellular sodium accumulation followed by water influxes. The failure of the ATPase dependent Na+/H+ and Na+/Ca2+ pumps, the competition with H+ for these Na+ pumps, and accumulation of extracellular K+ and excitotoxins, lead to depolarization and further reinforce these ion shifts. The loss of membrane function increases the risk of osmotic lysis. This process is a major component of primary neuronal death.3,24,25 Several hours after a severe HI injury there is a secondary phase of intracellular edema that is associated with the development of infarction and seizures.3 Presumably this corresponds to a secondary energy failure following perinatal asphyxia.23
Intracellular Calcium Accumulation
Intracellular calcium levels rise in response to NMDA receptor stimulation, release of calcium from intracellular stores (as binding to the endoplasmic reticulum is energy dependent and acidosis favors release of mitochondrial calcium), and loss of energy dependent in-out pumps.26 Large increases in free cytosolic calcium are toxic. Accumulation of calcium activates a number of enzymes including lipases, proteases, and endonucleases and contributes to free radical and prostanoid production.27 This process is thought to be a major component of primary and secondary injury.
Free Radical Formation
Free radicals are produced by several mechanisms particularly during reoxygenation28—these include oxidation of arachidonic acid, derivatives of purine oxidation, and the release of nitric oxide (NO).29,30 There are endogenous scavenger mechanisms involving the enzymes superoxide dismutase, glutathione peroxidase and catalase and damage occurs when the scavenger systems are overloaded. Nitric oxide, a potent vasodilator, can also be neurotoxic. Free radicals primarily act by attacking the fatty acid component of cell membranes (lipid peroxidation).2Q,31 NO also stimulates glutamate release.32 This process is primarily operative in the reperfusion phase— however, as macrophages also release free radicals, they may also play a role in delayed cell death.
Loss of Endothelial Integrity
Loss of endothelial integrity may exacerbate an asphyxial injury. Perhaps by allowing macrophage activation and invasion as well as entry of other vascular factors into the neural interstitium. The role of endothelial integrity, free radical damage and adhesion molecules such as the integrins are a focus of current research.
Excitotoxins
Glutamate activates at least three classes of membrane receptor: the NMDA, kainate and quisqualate receptors. The kainate receptor is linked to a sodium channel and the NMDA receptor complex includes a calcium channel. There are ontogenic changes in the relative concentrations of kainate and NMDA receptors.33,34 Ion fluxes are probably the major mechanism by which excessive glutamate leads to neuronal death.35,36 Extracellular glutamate accumulates during some forms of hypoxic-ischemic injury. This is due to the excessive depolarization of glutaminergic neurones and loss of energy dependent uptake and metabolism in glia.37-39 Thus excitoxicity is implicated both in primary neuronal injury and in secondary neuronal death where it is associated with delayed hyperexcitability or seizures.40-42
The Role of Seizures
Postischemic seizures lasting more than 30 minutes are associated with a poor prognosis43,44 and cerebral infarction.45 This is particularly the case in the low birth weight infant.46 There has been debate as to whether this is a causal association. Recently, we have shown, using an anticonvulsant dose of MK-801 6 hours after the injury to abolish postasphyxial seizure activity in the late gestation fetal sheep, that neuronal loss is significantly reduced particularly at sites distal to the presumed ictal focus in the region of cortical infarction.42
Macrophage and Microglial Activation
Activated microglia can be detected within an hour of global ischemia but increased activation occurs over several days. In addition with loss of endothelial integrity secondary to asphyxia, there can be an influx of bloodborn macrophages. These two processes can contribute markedly to delayed cell death.47 Activated macrophages and glia release a number of cytotoxins including NO, hydrogen peroxide48-50 and quinolinic acid.51 In addition they secrete cytokines49,52 which aggravate the cascade and also may promote scar formation. However it is not yet clear to what extent they contribute to secondary damage after HI injury in the developing brain.
Apoptosis
There is increasing evidence that some injuries can activate programmed or apoptotic cell death. Growth factors, protein synthesis and endonuclease inhibitors can ameliorate cell death in vitro. Further there is evidence that IGF-1, which can inhibit apoptosis in vitro, can reduce neuronal injury when administered after the insult.4 A number of markers of apoptosis such as fragmentation of DNA can be recognized after some brain injuries.53,54 If interference with apoptosis is the major mode of action of IGF-1 then apoptosis must be a component of delayed cell death.
Endogenous Protective Mechanisms
In addition to the destructive mechanisms described above it is necessary to consider the mechanisms the brain may use to ameliorate the injury as this also may suggest mechanisms of intervention.
Inhibitory Neuromodulators
Postasphyxial depression is a reflection of the release of a number of inhibitory modulators including adenosine, GABA, and somatostatin. It is probable that the postasphyxial depression is an endogenous response to reduce metabolic demand55 and thus helps recovery from the initial injury.
Hypothermia
The brain tends to tool spontaneously following injury which may be protective. After asphyxia neonates show decreased cerebral temperature which is thought to arise from reduced cerebral blood flow and metabolism.56 The spontaneous tooling of the brain that tends to occur during and after HI injury is neuroprotective in adult animals.57,58 Similarly preliminary studies in the infant rat suggest that maintaining the rat in a warm (32C°) environment after a moderate HI injury can markedly worsen histological outcome (unpublished).
Immune Modulators
The activation of the immune system after asphyxial brain injury is also accompanied by the expression of TGFß, an inhibitory immune modulator.59 Other factors are also likely to be expressed presumably as part of the process of limiting the extent of the inflammatory response.60 We have shown TGFß to be neuroprotective when given after an asphyxial injury61—limited evidence suggests an associated reduction in macrophage/microglial activation.
Neurotrophic Factors
It has been known for some years that bioassayable neurotrophic activity is found in the exudate of brains after brain injury; the level of activity is much higher in the neonate than the adult.62 We have used in situ hybridization to explore the basis of this increase after asphyxial injury. We were able to show that neither nerve growth factor or BDNF were expressed in the region of injury, and only minor expression was observed in the non-injured contralateral hippocampus secondary to postasphyxial seizures. In contrast, there is marked expression of IGF-1 and two of its binding proteins BP-2 and BP-3 in the region of injury.4,63 The IGF-1 is expressed by astrocytes within 1 to 3 days of injury. IGF-1 is a potent neurotrophin, which we have shown can reduce neural loss following central administration post injury.4 In contrast, IGF-2 is not expressed until some days later and BP-4 is inhibited. There is also evidence of increased expression of basic fibroblast growth factor but the extent of expression is much less.
While the mechanisms of action of this neurotrophic response are not definitively proven, it seems likely that IGF-1 acts by inhibiting apoptosis. IGF-1 is known to inhibit apoptosis in neuronal culture.
Microvascular Factors
Little is currently known of the endogenous vascular response after injury. However calcitonin gene related peptide (CGRP) which is a potent vasodilator is markedly expressed around small vessels in the brain after injury.64
Concluding Remarks
The above review illustrates some of the issues that need to be considered in addressing therapeutic approaches to asphyxia. The key factors that will need to be considered if clinical trials are planned include: definition of the time of intervention to the timing or phase of the injury, the role of sensitizing factors and the nature of the injury. The former will be the key factor in considering which potential mechanisms might be addressed therapeutically. Stratagems that address primary neuronal death are obviously only appropriate as prophylactic therapies. Those that address mechanisms operative in the reperfusion phase must be given before or during the insult. Perhaps the greatest promise lies in addressing the phenomenon of delayed cell death. At the present stage of knowledge it would appear that the most useful stratagems for neuronal rescue would be the use of neurotrophic agents that may arrest apoptosis, immunomodulators which may reduce the inflammatory response and suppression of post-asphyxial seizures.
Acknowledgments
The authors' work is supported by the Health Research Council of New Zealand.
References
- Gluckman PD, Williams CE. When and why do brain cells die? Dev Med Child Neurol 34:1010-1014, 1992.
- Massarweh WF, Vinters H, Schwartz P et al. Delayed neuronal necrosis in neonatal hypoxia-ischemia. Soc Neurosci Abstr 13:10, 1987.
- Williams CE, Gunn AJ,Gluckman PD. Time course of intracellular edema and epileptiform activity following prenatal cerebral ischemia in sheep. Stroke 22:516-521, 1991.
- Gluckman PD, Klempt ND, Guan J et al. A role for IGF-1 in the rescue of CNS neurons following hypoxic-ischemic injury. Biochem Biophys Res Commun 182:593-599, 1992.
- Guan J, Williams C, Gunning M et al. The effects of IGF-1 treatment after , hypoxic-ischemic brain injury in adult rats. J Cereb Blood Flow Metab 13:609-6 16,1993.
- Gunn AJ, Mydlar T, Bennet L et al. The neuroprotective actions of a calcium channel antagonist, flunarizine, in the infant rat. Pediatr Res 25:573-576, 1989.
- Gunn AJ, Gluckman PD. Flunarizine, a calcium channel antagonist, is not neuroprotective when given after hypoxia-ischemia in the infant rat. Dev Pharmacol Ther 17:205-209, 1991.
- Mallard EC, Gunn AJ, Williams CE et al. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol 167:1423-1430, 1992.
- Mallard EC, Williams CE, Johnston BM, Gluckman PD. Increased vulnerability to
neuronal damage following umbilical cord occlusion in the fetal sheep with advancing gestation. Am J Obstet Gynecol 170:206-214, 1994. - Hansen A. Extracellular potassium concentration in juvenile and adult rat brain cortex during anoxia. Acta Physiol Scand 99:412-420, 1977.
- Cook CJ, Gluckman PD, Williams CE, Bennet L. Precocial neural function in the growth retarded fetal lamb. Pediatr Res 24:600-605, 1988.
- Pettigrew AG, Edwards DA, Henderson-Smart DJ. The influence of intrauterine growth retardation on brainstem development of preterm infants. Dev Med Child Neurol
27:467-472, 1985. - Thordstein M, Kjellmer I. Cerebral tolerance of hypoxia in growth-retarded and
appropriately grown newborn guinea pigs. Pediatr Res 24:633-638, 1988. - Voorhies TM, Rawlinson D, Vannucci RC. Glucose and perinatal hypoxic-ischemic brain damage in the rat. Neurology 36:1115-1118, 1986.
- Reeves I, Mujsce D, Vannucci RC. Extreme hyperglycemia protects the perinatal brain from hypoxic-ischemic damage. Pediatr Res 31:352A, 1992.
- Jakubovicz D, Klip A. Lactic acid-induced swelling in C6 glial cells via Na+/ H+ exchange. Brain Res 485:215-224, 1989.
- Giffard RG, Monyer H, Christine CW, Choi DW Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res 506:339-342 1990.
- Tombaugh GC, Sapolsky RM. Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res 506:343-345, 1990.
- Dietrich WD. The importance of brain temperature in cerebral injury. J Neurotrauma 9:S475-S485, 1992.
- Gunn AJ, Parer JT, Mallard EC et al. Cerebral histological and electrophysiological changes after asphyxia in fetal sheep. Pediatr Res 31:486-491, 1992.
- Williams CE, Gunn AJ, Mallard EC, Gluckman PD. Outcome after ischemia in the developing sheep brain: An electroencephalographic and histological study. Ann Neurol
31:14-21, 1992. - Mallard EC, Williams CE, Gunn AJ et al. Frequent episodes of brief ischemia sensitize the fetal sheep brain to neuronal loss and induce striatal injury. Pediatr Res 33:61-65,1993.
- Wyatt JS, Edwards AD, Azzopardi D, Reynolds EO. Magnetic resonance and near infrared spectroscopy for investigation of perinatal hypoxic- ischaemic brain injury. Arch Dis Child 64:953-963, 1989.
- Schuier F, Hossmann KA. Experimental brain infarcts in cats II. lschemic brain edema. Stroke 11:593-601, 1980.
- Choi DW. Cerebral hypoxia: Some new approaches and unanswered questions. J Neurosci 10:2493-2501, 1990.
- Siesjö BK, Bengtsson F, Grampp W, Theander S. Calcium, excitotoxins, and neuronal death in the brain. Ann NY Acad Sci 568:234-251, 1989.
- Siesjö BK, Ekholm A, Katsura K et al. The type of ischemia determines the
pathophysiology of brain lesions and the therapeutic response to calcium channel blockade. In: Krieglstein J, Oberpichler H, (eds). Pharmacology of Cerebral lschemia. Stuttgart: issenschaft-liche Verlagsgesellscaft, pp. 79-88, 1990. - Goplerud JM, Prakash MO, Delivoria-Papadopoulos M. Brain cell membrane dysfunction following acute asphyxia in newborn piglets. Biol Neonate 61:33-41, 1992.
- Siesjö BK. Cell damage in the brain: A speculative synthesis. J Cereb Blood Flow Metab 1:155-167, 1981.
- Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol 15:53-59, 1991.
- McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312:159-163, 1985.
- Giulian D, Vaca K, Noonan CA. Secretion of neurotoxins by mononuclear phagocytes infected with HIV-1. Science 250:1593-1596, 1990.
- Greenamyer JT, Penney JB, Young AB. Autoradiographic characterization of
N-methyl-D-aspartate, quisqualate and kainate-sensitive glutamate binding sites. J Pharmacol Exp Ther 233:254-263, 1985. - Greenamyre T, Penney J, Young A. Evidence for transient perinatal glutamatergic innervation of globus pallidus. J Neurosci 7:1022-1030, 1987.
- Siesjö BK, Bengtsson A. Calcium fluxes, calcium antagonists, and calcium-related pathology in brain ischemia, hypoglycemia, and spreading depression: A unifying hypothesis.J Cereb Blood Flow Metab 9:127-140, 1989.
- Meldrum B. Excitatory amino acids and anoxic/ischemic brain damage. TINS 8:47-48, 1985.
- Torp R, Andine P, Hagberg H et al. Cellular and subcellular redistribution of glutamate-,glutamine- and taurine-like immuno-reactivities during forebrain ischemia: A semiquantitative electron microscopic study in rat hippocampus. Neurosci 4 1:433-447, 1991.
- Sloper J, Johnson P, Powell TPS. Selective degeneration of interneurons in the motor cortex of infant monkeys following controlled hypoxia: A possible cause of epilepsy. Brain Res 198:204-209, 1980.
- Romijn H, Ruijter J, Wolters P. Hypoxia preferentially destroys GABAnergic
neurons in developing rat neocortex explants in culture. Exp Neurol 100:332-340,
1987. - Andine P, Orwar O, Jacobson I et al. Changes in extracellular amino acids and
spontaneous neuronal activity during ischemia and extended reflow in the CAl of the rat hippocampus. J Neurochem 57:222-229, 1991. - Hattori H, Morin AM, Schwartz PH et al. Posthypoxic treatment with MK-801
reduces hypoxic-ischemic damage in the neonatal rat. Neurology 39:713-718,
1989. - Tan WKM, Williams CE, Gunn AJ et al. Suppression of post-ischemic
epileptiform activity with MK-801 improves neural outcome in fetal sheep. Ann
Neurol 32:677-682, 1992. - Mellits E, Holden K, Freeman J. Neonatal seizures II. A multivariate analysis of
factors associated with outcome. Pediatrics 70:177-185, 1982. - Tudehope D, Harris A, Hawes D, Hayes M. Clinical spectrum and outcome of
neonatal convulsions. Aust Paediatr J 24: 249-253, 1988. - Williams CE, Gunn AJ, Gluckman PD, Synek B. Delayed seizures occurring with
hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res 27:561-565, 1990. - Watkins A, Szymonowicz W, Jin X, Yu VV. Significance of seizures in very
low-birthweight infants. Dev Med Child Neurol 30:162-169, 1988. - Giulian D, Robertson C. Inhibition of mononuclear phagocytes reduces ischemic injury in the spinal cord. Ann Neurol 27:33-42, 1990.
- Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mammalian brain. J Neurosci 6:2163-2178, 1986.
- Giulian D, Chen J, Ingeman JE et al. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci 9:4416-4429, 1989.
- Thery C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing neurons. Eur J Neurosci 3:1155-1164, 1991.
- Kohler C, Eriksson LG, Flood PR et al. Quinolinic acid metabolism in the rat brain.
Immunohistochemical identification of 3- hydroxyanthranilic acid oxygenase and quinofinic acid phosphoribosyltransferase in the hippocampal region. J Neurosci 8:975-987, 1988. - Giulian D, Baker TJ, Shih LC, Lachman LB. Interleukin 1 of the central nervous system is produced by ameboid microglia. J Exp Med 164:594- 604, 1986.
- Okamoto M, Matsumoto M, Ohtsuki T et al. Apoptosis as an underlying mechanism of delayed neuronal death. J Cereb Blood Flow Metab 13:Abstract XI-12-S 80, 1993.
- Raff MC. Social controls on cell survival and cell death. Nature 356:397-400, 1992.
- Leffler CW, Busija DW, Mirro R et al. Effects of ischemia on brain blood flow and oxygen consumption of newborn pigs. Am J Physiol 257H:1917 -1926, 1989.
- Weninger M, Simbruner G, Malamitsi Puchner A. Heat flux from the head surface in healthy newborns and in newborns with cerebral pathology. Wen KlinWochenschr 101:529-533, 1989.
- Boris-Möller F, Smith M-L, Siesjö BK. Effects of hypothermia on ischemic brain damage: A comparison between preischemic and postischemic tooling. Neurosci Res Comm 5(2):87-94, 1989.
- Moyer DJ, Welsh FA, Zager EL. Spontaneous cerebral hypothermia diminishes focal infarction in rat brain. Stroke 23:1812-1816, 1992.
- Klempt ND, Sirimanne E, Gunn AJ et al. Hypoxia-ischemia induces transforming growth factor-beta mRNA in the infant rat brain. Mol Brain Res 13:93-101, 1992.
- Berkenbosch F. Macrophages and astroglial interactions in repair to brain injury. Ann NY Acad Sci 650:186-190, 1992.
- McNeill H, Guan J, Miller O et al. Neuronal rescue with transforming growth factor-beta 1 after hypoxic-ischemic brain injury. Neuro Report 5:901-904, 1994.
- Nieto-Sampedro M, Lewis ER, Cotman CW et al. Brain injury causes a time-dependent increase in neurotrophic activity at the lesion site. Science 217:860-861, 1982.
- Klempt ND, Klempt M, Gunn AJ et al. Expression of insulin-like growth factor binding protein-2 (IGFBP-2) following transient hypoxia-ischemia in the infant rat brain. Brain Res 15:55-61, 1992.
- Dragunow M, Sirimanne E, Lawlor PA et al. Accumulation of calcitonin-generelated peptide-like immunoreactivity after hypoxic-ischaemic brain injury in the infant rat. Mol Brain Res 14:267-272, 1992.
Fetal Brain Metabolism Under Stress—Oxygenation, Acid-Base and Glucose
Julian T. Parer, M.D., Ph.D.
Cardiovascular Research Institute and Department of Obstetrics, Gynecology and Reproductive Sciences, University of California, San Francisco
Introduction
Cerebral oxidative metabolism is well described in fetal sheep at two stages of development, and is known to remain relatively constant over a wide range of oxygen content in arterial blood. This constancy of oxygen consumption is due to an increase in cerebral blood flow which matches the reduction in oxygen content and oxygen extraction. Although a number of factors are involved in the hypoxia-associated vasodilation (e.g., O2, CO2, adenosine, prostaglandins, arginine vasopressin, etc.) its regulation is incompletely understood. During severe asphyxia, however, there is a limit to the vasodilator function, and both cerebral blood flow and oxygen consumption fall. The fetus can tolerate a certain degree of reduced oxygen uptake (possibly to 50 percent of control), by various conservation techniques, but severe reductions are associated with neuronal damage.
The primary substrate for the fetal brain under normal conditions is glucose, but the fetus can readily use anaerobic glycolysis and produce lactate under conditions of oxygen limitation. Lactate efflux from the brain is relatively slow, so prolonged and severe asphyxia may result in a high tissue level, which has been implicated in neuronal damage.
Fetal Brain Metabolism Under Conditions of Normal Oxygenation
Normal Values
Oxygen consumption. Most measurements of fetal brain cerebral cortex oxygen consumption have been obtained in fetal sheep, with the modified Fick principle. Blood oxygen content is measured with catheters in the preductal (ascending) aorta and sagittal sinus, and cerebral cortical blood flow is measured using the radionuclide microsphere technique. In the near term fetal sheep the mean cerebral blood flow is 120 m1/100g/min, and arteriovenous oxygen content difference 1.6 mM. This results in a mean cerebral oxygen consumption of about 190 mM/100g/min.1
The oxygen consumption is similar to this in the adult sheep and newborn.2 However, in the fetus the oxygen delivery is 70 percent greater than that in the adult, implying an excessive blood flow for the resting needs. Thus the fetal cerebral blood flow is higher than the adult at any arterial oxygen content. This may be a physiologic adaptation to the lower PO2 in the fetal blood, or a reserve mechanism for anticipated stressful conditions.3
In adults and newborns it is estimated that approximately half of the oxygen consumption supplies energy for synaptic transmission. One- quarter of the oxygen consumption maintains neuronal membrane potentials, and a further one-quarter isdevoted to unidentified processes.2
Carbohydrate metabolism. Glucose consumption in the near term fetal sheep is approximately 26 mM/100g/min. The oxygen-glucose index, which is a measure of the extent to which complete metabolism of the glucose can explain the total oxygen uptake, is 100 percent in such sheep, suggesting that glucose is the major and possibly only substrate used by the brain under normal conditions. However, recent work by Chao et al.4 points to a portion of the energy during the low voltage state (with higher metabolic rates) being supplied anaerobically from glucose.
Developmental Changes
Oxygen consumption in fetal sheep at 93 days (0.63 of gestation) is approximately 50 mM/l00g/min, that is 25 percent of the value in the near term fetus.5 This reduced metabolism occurs in the face of a reduced cerebral cortical blood flow rather than a reduction in fractional oxygen extraction or arteriovenous oxygen content difference across the brain. This lower O2 uptake may reflect less developed synaptic activity, and may also be a consequence of the lesser mitochondrial mass in the immature brain.5
In the sheep fetus at 0.63 of gestation the cerebral glucose consumption is 8.5 mM/100g/min, i.e., 30 percent of that in the near term sheep fetus. There is net lactate production under normal conditions, which can explain a further 15 percent of the glucose utilization, and together with the glucose, essentially all of the oxygen consumption. Because of the high oxygen values in fetal sheep blood there is no reason to believe lactate is a result of insufficient oxygen availability.5
Influence of Fetal State
The fetal sheep near term spends the most time in the low voltage state, during which time rapid eye and rapid irregular breathing movements occur. Measurements in chronically prepared fetal sheep show that during the high voltage state the cerebral oxygen consumption is 83 percent of that found in the more active low voltage state. The difference may represent increased brain neuronal activity or an increase in synthesis within the brain in the low voltage state.6,7
As with oxygen consumption, the glucose consumption in the fetus is also increased during low-voltage electrocortical activity8,9 (see section on Carbohydrate metabolism).
Glucose consumption has been shown to be dependent on auditory input in the near term fetal sheep. In fetuses with cochlear ablation, using the C14 deoxyglucose method, local glucose utilization was depressed in most of the gray and white matter examined, and was reduced 25 percent in brain stem auditory nuclei.10 Furthermore, glucose utilization in many cerebral structures was elevated in noise exposed fetuses.11
Regulation of Fetal Brain Metabolism
In the adult brain there is acceptance of the concept that local cerebral blood flow is normally distributed in almost the exact proportion to the rates of glucose utilization, and that the blood flow and local glucose consumption change in response to local functional activity.12 This coupling is relatively poorly studied in the sheep under normal conditions, but more is known under pathologic perturbations.
Variations in cerebral cortical blood flow can be due to variations in oxygen content and carbon dioxide levels of arterial blood. Even within the normal range blood flow increases as oxygen decreases, resulting in a constant cerebral oxygen consumption.1,13 Szymonowicz et al.,14 however, concluded that cerebral blood flow was not primarily determined by O2 content when variations occurred within the physiological range. In addition to O2 control, it is known that cerebral blood flow increases as CO2 tension increases.
Autoregulation of cerebral blood flow occurs in the adult and also in the fetus.15,17,18 Thus there is a range of arterial blood pressure over which cerebral blood flow remains stable. It has been shown that in the preterm lamb the range is narrowed, compared to the term lamb, and that the mean resting carotid arterial blood pressure lies close to the lower limit of autoregulation.18
Fetal Brain Metabolism During Asphyxia
Asphyxia
Asphyxia is best described as insufficiency of exchange of the respiratory gases. Fetal asphyxia almost always occurs as a result of insufficient umbilical or insufficient uterine blood flow. While this definition begs the question of what is insufficient, it is recognized that reduction of these blood flows below a certain level will result in reduction of oxygen delivery to the fetus, and potentially to the brain, and this could result in reduced oxygen consumption by that organ. Under these conditions anaerobic metabolism may be utilized for high energy bond production, and lactate will be the end product. This will produce a metabolic acidosis in the tissue. At the same time there may be insufficient removal of carbon dioxide from the tissues, and a concomitant respiratory acidosis will develop.
The definition of asphyxia thus includes a reduction in oxygen content, an elevation of PCO2, and a reduced pH. The definition of insufficiency can depend on arbitrary values of these three variables (e.g., mean ± 1 S.D.), or it may depend on a measurement of oxygen consumption falling significantly below the mean normal value. None of these criteria are particularly valuable for defining when there is permanent loss of function. Nevertheless, it is of value to examine fetal responses to asphyxia in order to determine compensatory mechanisms.
Fetal Oxygen Consumption During Induced Hypoxia/Asphyxia
During hypoxia or asphyxia produced in the fetus by various techniques, there is a decrease in cerebral vascular resistance and an increase in cerebral blood flow.1,15,19-23 The increase in blood flow is such that the oxygen consumption of the cerebral hemispheres remains constant over the range of ascending aortic oxygen tensions of 14-36 mm Hg.1 CaO2 has the best overall correlation with cerebral blood flow among different types of hypoxia.2 In addition to the role of decreased oxygen in bringing about this vasodilatation, increasing carbon dioxide tension is also involved in vasodilation of the cerebral vascular bed during asphyxia.15,16
In the mid term fetal sheep there is an increase in cerebral blood flow during hypoxia but this is less than that seen in the term fetus, so that oxygen consumption of the brain was maintained by combined increased fractional oxygen oxtraction and increased blood flow.24 The authors suggested that this may have been due to immature regulatory mechanisms.
As noted above autoregulation has been demonstrated in the fetal lamb, such that blood flow to the brain is maintained nearly constant over a wide range of arterial pressures. This autoregulation has been shown to be dependent on an adequate level of arterial oxygen, because during hypoxia cerebral blood flow became pressure dependent.17
Under conditions of severe asphyxia when uterine blood flow was 25 percent of control, it was found that sufficient augmentation of the cerebral blood flow was no longer maintained, and similar values to control were obtained.25 There was a doubling of the vascular resistance in the cerebral vasculature compared to normoxic control values, and a further increase in arterial blood pressure. This decrease in blood flow, coupled with a decreased arteriovenous oxygen difference during more profound hypoxemia, results in a decrease in cerebral oxygen consumption to as much as half of normal.26
This reduced consumption appears to be proportional to the degree of hypoxemia as measured by arterial oxygen content (Figure 1), and is due to the fact that cerebral vascular resistance does not decrease further in response to and in proportion to the increasing hypoxemia. Thus cerebral blood flow can no longer be augmented below a certain level of hypoxemia, and with the progressive obligatory reduction in arteriovenous oxygen difference, the uptake of oxygen falls. The reduction in cerebral oxygen consumption appears to occur when ascending aorta oxygen content is below 1 mM.
A decrease in cerebral oxygen consumption was also demonstrated to occur after 7.2 hours of isocapnic hypoxia in fetal sheep when the arterial oxygen content was below 0.8 mM.27
The inability of the fetus to maintain sufficient oxygen delivery to the brain had previously been predicted on the basis of the increased fraction of cardiac output (from 25 to 50 percent) required to be directed towards the heart and central nervous system.28
FIGURE 1: Cerebral Oxygen Consumption (vcO2) in Fetal Sheep Related to Ascending Aortic Oxygen Content (CaO2). y=[(- 3.91/x) + 13.3]2
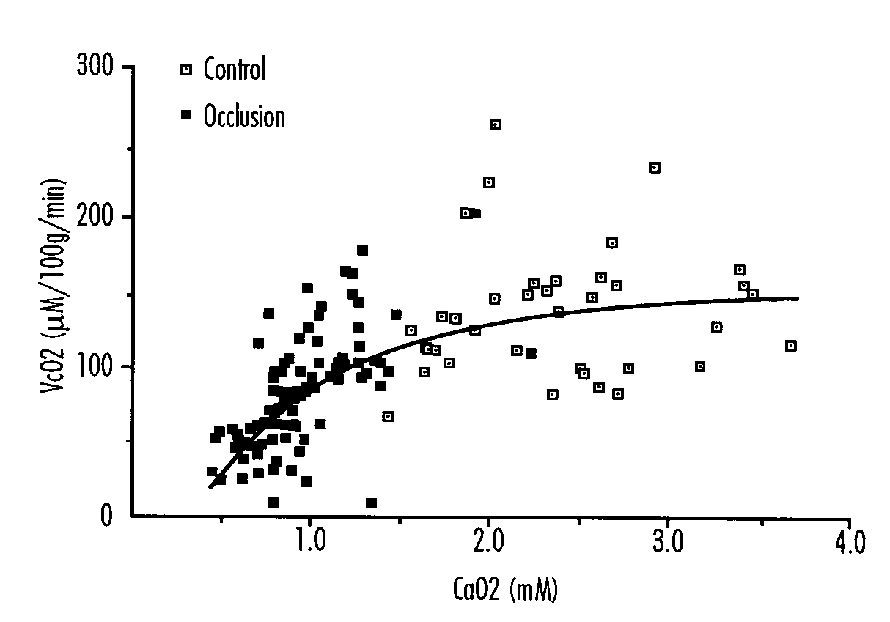
These authors on the basis of mathematical modeling suggested that when ascending aortic oxygen content was reduced from 1 to 0.5 mmol/l-1 such a compensation could not take place, and the cardiovascular system may begin to fail in delivering adequate amounts of oxygen to at least some parts of the central nervous system.
Carbohydrate Metabolism During Asphyxia
During hypoxia/asphyxia of moderate to severe degrees the circulating glucose concentration rises by approximately 50 percent in fetal sheep. Similarly there is development of a metabolic acidosis, most of which can be explained by increased lactate levels.29
The glucose and lactate flux across the brain has been studied in the fetal sheep during cerebral ischemia produced by partial occlusion of the brachiocephalic artery.9 During severe ischemia there is reduced brain oxygen consumption, approximately 26 percent, and increased glucose uptake, approximately 25 percent. This is considerably more glucose than can account for the oxygen uptake. The brain lost lactate during occlusion, but not sufficient to explain anaerobic metabolism of the glucose. The authors concluded that lactate accumulated in the brain tissue because of inability of the blood-brain barrier to transport it, and that this may contribute to brain injury, in which elevated lactate levels have been previously implicated in adult and immature individuals.
During combined hypoxemia and cerebral ischemia, however, the authors could not detect a net lactate flux.30 They suggested that this may be due to a concomitant cerebral and systemic increase in lactate concentration.
In a similar model, it was shown that glucose infusion tended to maintain electroencephalographic amplitude during cerebral ischemia, thus suggesting it had a protective effective.31 In further studies, fetal glucose levels were reduced 33 percent by insulin infusion.32 This did not produce any short-term reduction in cerebral oxygen or glucose consumption.
The presence of adequate brain carbohydrate stores has been demonstrated in the past to be an important determinant of tolerance to asphyxia at birth. Thus, the survival time of insulin treated newborn rats was only one tenth that of normoglycemic litter mates when exposed to nitrogen.33
Reduced Cerebral Metabolism and Neuronal Damage
There is relatively little information about the relationship between reduced cerebral oxygenation and neuronal cell damage. We have reported cerebral histologic and electrophysiologic changes after asphyxia in chronically instrumented fetal sheep, induced by reducing uterine blood flow to result in an ascending aortic blood oxygen content < 1.5 mM.29 In an initial protocol, asphyxia continued for up to 60 minutes, and in a subsequent study supplementary maternal hypoxia was added if full occlusion of the common uterine artery for 15 minutes did not reduce the EEG voltage to less than 20 percent of baseline.
Uterine artery occlusion resulted in severe hypoxemia, hypercarbia, acidosis and an initial hypertension and bradycardia. Eight of 14 surviving fetuses showed neuronal damage with greatest loss in the parasagittal cortex, striatum, and the CA2 region of the hippocampus, after 3 days. Neuronal damage was strongly associated with the minimum blood pressure during the insult but not with the degree of hypoxia. No other factor was independently predictive, but when considered separately the pH at the end of asphyxia and loss of intensity of the EEG were also correlated with outcome. The pH fell to < 7.0 in 6 of 8 with damage while it remained > 7.0 in 5 of 6 with no damage (p<0.05). We concluded that severe intrauterine asphyxia for periods of 30 to 120 minutes can cause predominantly parasagittal neuronal death, and that this is associated with hypotension, severe metabolic acidosis and suppression of EEG during the insult.29 These data are consistent with the suggestion that impairment of cerebral perfusion is a critical event in causing cerebral damage during perinatal asphyxia.34
We have measured cerebral oxygen consumption in a further series of fetal sheep using the above protocol.35 In the animals that subsequently developed seizure activity, the fetal arterial oxygen content (CaO2) fell from 3.0 ± 0.9 mM (mean ± SD) to 0.5 mM. The blood flow to the cerebral cortex during control was 163 ± 51 ml/min/l00g, and increased to 90 percent above control by 30 minutes of asphyxia. It then progressively fell to approach control values by 90 minutes. The arteriovenous O2 difference narrowed so that cortical O2 consumption decreased to 36 percent of control. The fetal arterial pH fell from 7.39 ± 0.03 to 6.89 ± 0.01, the base excess from 4.7 ± 2.4 mEq/1 to -21.6 ± 5.7 mEq/1, and the PCO2 rose from 49 ± 3 mm Hg to 66 ± 0 mm Hg, during the asphyxial insult. Fetuses that survived without seizures generally had lower falls in cortical O2 consumption. Fetuses that died during or shortly after the insult either had arrhythmias or a rapid progression of asphyxia. These data suggest that depression of O2 consumption by the fetal cortex to less than 50 percent of control over approximately 90 minutes results in neurologic damage as demonstrated by seizures. Damage to other organs was apparently not sufficient to be lethal within 24 hours.35
With respect to another technique for producing fetal asphyxia, Mallard et al.36 have produced neuronal cell loss in the hippocampus by severe umbilical cord occlusion for 10 minutes in near term fetal sheep. Although the duration was short, there was severe asphyxia, hypoxemia, bradycardia and electrocorticographic suppression for up to 5 hours. Three of 17 animals did not survive the asphyxia. The metabolism during asphyxia was not quantitated, but it was most likely severely depressed. We have produced seizures after umbilical cord occlusion of lesser severity and for a longer duration.37
We do not have data on the threshold of reduction in oxygen consumption that causes damage to the fetal brain. It seems likely, however, that a 15 percent reduction would be tolerable. It has been shown that a change in electrocorticographic activity from low voltage high frequency to high voltage low frequency activity is associated with a similar decrease in oxygen uptake.6 The degree of hypoxemia seen in our moderately asphyxiated fetuses is usually associated with such an electroencephalographic change from low to high voltage, and this alone may explain the decrease in cerebral oxygen consumption.
The reduction of cerebral oxygen consumption to approximately 50 percent of control may be associated with further compensatory mechanisms for preventing neuronal damage, but we cannot determine such from our studies. Severe asphyxia in the immediate newborn period has been associated with functional and anatomic central nervous system damage in monkeys,38 but it is not possible to compare the physiologic conditions of that study with those quoted above.
These studies have some important clinical implications. They show the remarkable conservation strategies available to the fetus despite quite substantial hypoxemia, mainly due to the fetal capacity for augmenting blood flow. This may explain why intact survival is not infrequently seen in the human fetus despite profound documented asphyxia at birth.39-41 With profound asphyxia however, there is decompensation of these mechanisms and such fetuses may subsequently develop hypoxic neuronal damage.
Regulation of Cerebral Metabolism During Asphyxia
The fetal brain blood flow is sensitive to changes in O2 and CO2, and as noted, metabolic rate is constant over a wide range of O2 content of perfusing blood, because there is a compensatory balance between the blood flow and arteriovenous oxygen concentration difference. The cerebral blood flow is also directly proportional to PCO2, but this almost certainly does not result in increased oxygen consumption by the brain. The response may teleologically be thought of as a mechanism for reducing elevated brain CO2.
The mechanisms of the increased brain blood flow in response to either of these mechanisms are unsure.CO2 has an independent effect, and this may alter the brain's ability to tolerate hypoxia, because as CO2 falls, brain blood flow also falls, and in order to maintain oxygen consumption, oxygen extraction must increase. During hypoxia this may be limited, so oxygen consumption may fall.3 This is of less importance in the fetus because during asphyxia in utero CO2 almost invariably rises, unless there is extreme maternal hyperventilation.
There are several possible mechanisms for the variations in cerebral blood flow during hypoxia, and one possibility (extrapolating from adult studies) is a direct action of oxygen tension on smooth muscle. Oxygeneses have been suggested as O2 sensors in mediating the responses.42 Release of the vasodilator adenosine may be one such mechanism.43 It has been shown that brain vascular resistance increases and brain blood flow decreases during hypoxia in fetal sheep in response to arginine vasopressin44 and prostaglandin45 blockade, thus demonstrating a role for these substances. There may also be other as yet unidentified substances.
Miscellaneous Factors Affecting Cerebral Metabolism
Anesthesia and Drugs
Pentobarbital resulted in a 27 percent decrease in cerebral oxygen consumption at constant perfusion pressure in normoxemic fetal sheep.46 During hypoxia, with barbiturate treatment, there were still increases in cerebral blood flow, but in proportion to the reduced metabolism.47 Similar studies with both halothane48 and isofluorane-oxygen49 in fetal sheep demonstrated retention of the ability of the fetal cerebral circulations to vasodilate during asphyxia.
Ethanol infusion resulted in a 23 percent reduction of cerebral oxygen consumption in near term fetal sheep.50 The dosage was selected to mimic episodic binge type drinking in humans. The authors concluded that the reduced cerebral metabolism represented a direct depressant effect on tissue (e.g., protein or DNA) synthesis and an indirect effect on fetal states. This may explain growth abnormalities in infants exposed to large doses of alcohol in utero.
In contrast to this, alcohol administration to more immature sheep fetuses had little influence on cerebral oxidative or carbohydrate metabolic rates.51 The difference between the responses of the mature and immature fetuses remains unexplained.
Status Epilepticus
Oxygen consumption by the brain is known to increase in the adult during generalized seizures. Local glucose cerebral metabolism has been studied in newborn primates, and increased up to fourfold to eightfold in the cortex.52 The latter authors noted that after 45 minutes of seizures the glucose uptake fell twofold, probably because energy demand exceeded glucose supply.
Increased Intracranial Pressure
Increased intracranial pressure caused by infusion of fluid into the lateral ventricle caused an increase in arterial blood pressure, and maintenance of perfusion pressure, which resulted in maintenance of cerebral blood flow.53 There was a concomitant decrease in visceral blood flow. There was thus maintenance of cerebral oxygen consumption during this simulated head compression, possibly mediated by increases in epinephrine, norepinephrine and arginine vasopressin.
References
- Jones MD, Jr, Sheldon RE, Peeters LL et al. Fetal cerebral oxygen consumption at different levels of oxygenation. J Appl Physiol: Respirat Environ Exercise Physiol 43:1080, 1977.
- Jones MD, Jr, Traystman RJ. Cerebral oxygenation of the fetus, newborn, and adult. Semin Perinatol 8:205, 1984.
- Jones MD, Jr, Rosenberg AA, Simmons MA et al. Oxygen delivery to the brain before and after birth. Science 216:324, 1982.
- Chao CR, Hohimer AR, Bissonnette JM. The effect of electrocortical state on cerebral carbohydrate metabolism in fetal sheep. Dev Brain Res 49:1, 1989.
- Gleason CA, Hamm C, Jones MD, Jr. Cerebral blood flow, oxygenation, and carbohydrate metabolism in immature fetal sheep in utero. Am J Physiol 256:R1264, 1989.
- Richardson BS, Patrick JE, Abduljabbar H. Cerebral oxidative metabolism in the fetal lamb: Relationship to electrocortical state. Am J Obstet Gynecol 153:426, 1985.
- Richardson BS. The effect of behavioral state on fetal metabolism and blood flow circulation. Semin Perinatol 16:227, 1992.
- Abrams R, Hutchison AA, Jay TM, Kennedy C. Cerebral glucose utilization increased during rapid eye movement sleep. Ann Neurol 20:401, 1986.
- Chao CR, Hohimer AR, Bissonnette JM. Cerebral carbohydrate metabolism during severe ischemia in fetal sheep. J Cerebral Blood Flow Metab 9:53, 1989.
- Abrams RM, Hutchison AA, McTiernan MJ, Merwin GE. Effect of cochlear ablation on local cerebral glucose utilization in fetal sheep. Am J Obstet Gynecol 157:1438, 1987.
- Abrams RM, Hutchison AA, Gerhardt KJ et al. Local cerebreal glucose utilization in fetal sheep exposed to noise. Am J Obstet Gynecol 157:456, 1987.
- Sokoloff L. Relationships among local functional activity, energy metabolism, and blood flow in the central nervous system. Federation Proc 40:2311, 1981.
- Jones MD, Jr, Sheldon RE, Peeters LL et al. Regulation of cerebral blood flow in the ovine fetus. Am J Physiol 235:H162, 1978.
- Szymonowicz W, Walker AM, Cussen L et al. Developmental changes in regional cerebral blood flow in fetal and newborn lambs. Am J Physiol 254:H52, 1988.
- Purves MJ, James IM. Observations on the control of cerebral blood flow in the sheep fetus and newborn lamb. Circ Res 25:651, 1969.
- Rosenberg AA, Jones MD, Jr, Traystman RJ et al. Response of cerebral blood flow to changes in PCO2 in fetal, newborn, and adult sheep. Am J Physiol 242:H862, 1982.
- Tweed WA, Cote J, Pash M, Lou H. Arterial oxygenation determines autoregulation of cerebral blood flow in the fetal lamb. Pediatr Res 17:246, 1983.
- Papile L-A, Rudolph AM, Heyman MA. Autoregulation of cerebral blood flow in preterm fetal lamb. Pediatr Res 19:159, 1985.
- Cohn HE, Sacks EJ, Heymann MA, Rudolph AM. Cardiovascular responses to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol 120:817, 1974.
- Peeters LLH, Sheldon RE, Jones MD et al. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol 135:637, 1979.
- Johnson GN, Palaniuk RJ, Tweed WA et al. Regional cerebral blood flow changes during severe fetal asphyxia produced by slow partial umbilical cord compression. Am J Obstet Gynecol 135:48, 1979.
- Ashwal S, Majcher JS, Van N, Longo LD. Patterns of fetal lamb regional cerebral blood flow during and after prolonged hypoxia. Pediatr Res 14:1104,1980.
- Reuss ML, Parer JT, Harris JL, Krueger TR. Hemodynamic effects of alpha-adrenergic blockade during hypoxia in fetal sheep. Am J Obstet Gynecol 42:410, 1982.
- Gleason CA, Hamm C, Jones MD, Jr. Effect of acute hypoxemia on brain blood flow and oxygen metabolism in immature fetal sheep. Am J Physiol 258 (Heart Circ Physiol 27):H1064, 1990.
- Yaffe H, Parer JT, Block BS, Llanos AJ. Cardiorespiratory responses to graded reductions of uterine blood flow in the sheep fetus. J Dev Physiol 9:325, 1987.
- Field DR, Parer JT, Auslender RA et al. Cerebral oxygen consumption during asphyxia in fetal sheep. J Dev Physiol 14:131, 1990.
- Richardson BS, Rurak D, Patrick JE et al. Cerebral oxidative metabolism during sustained hypoxemia in fetal sheep. J Dev Physiol 11:37, 1989.
- Sheldon RE, Peeters LLH, Jones MD, Jr et al. Redistribution of cardiac output and oxygen delivery in the hypoxemia fetal lamb. Am J Obstet Gynecol 135:1071, 1979.
- Gunn AJ, Parer JT, Mallard EC et al. Cerebral histologic and electrocorticographic changes after asphyxia in fetal sheep. Pediatr Res 31:486, 1992.
- Hohimer AR, Chao CR, Bissonnette JM. The effect of combined hypoxemia and cephalic hypotension on fetal cerebral blood flow and metabolism. J Cerebral Blood Flow Metab 11:99, 1991.
- Chao CR, Hohimer AR, Bissonnette JM. The effect of elevated blood glucose on the electroencephalogram and cerebral metabolism during short- term brain ischemia in fetal sheep. Am J Obstet Gynecol 161:221, 1989.
- Bissonnette JM, Hohimer AR, Richardson BS, Machida CM. Effect of acute hypoglycaemia on cerebral metabolic rate in fetal sheep. J Dev Physiol 7:421, 1985.
- Vannucci RC, Vannucci SJ. Cerebral carbohydrate metabolism during hypoglycemia and anoxia in newborn rats. Ann Neurol 4:73, 1978.
- Ting P, Yamaguchi S, Bacher JD et al. Hypoxic-ischemic cerebral necrosis in midgestational sheep fetuses: Physiopathologic correlations. Exp Neurol 80:227, 1983.
- Ball RH, Espinoza MI, Parer JT et al. Cerebral blood flow and metabolism in ovine fetuses during asphyxia resulting in seizures. J Mat Fet Med 3:157-162, 1994.
- Mallard EC, Gunn AJ, Williams CE et al. Transient umbilical cord occlusion causes hippocampal damage in the fetal sheep. Am J Obstet Gynecol 167:1423, 1992.
- Ball RH, Parer JT, Caldwell LE, Johnson J. Regional blood flow and metabolism in ovine fetuses during severe cord occlusion. Am J Obstet Gynecol 171:1549-1555, 1994.
- Myers RE. Two patterns of perinatal brain damage and their conditions of occurrence. Am J Obstet Gynecol 112:246, 1972.
- Freeman JM, Nelson KB. Intrapartum asphyxia and cerebral palsy. Pediatrics 82:240, 1988.
- Paneth N, Stark R. Cerebral palsy and mental retardation in relation to indicators of perinatal asphyxia. Am J Obstet Gynecol 147:960, 1983.
- Low JM, Gailbraith RS, Muir DW et al. Motor and cognitive deficits after intrapartum asphyxia in the mature fetus. Am J Obstet Gynecol 43:1080, 1977.
- Traystman RJ, Gurtner GH, Rogers MC et al. A possible role of oxygeneses in the regulation of cerebral blood flow. Adv Physiol Sci 9:167, 1981.
- Rubio R, Berne RM, Bockmen EL et al. Relationship between adenosine concentration and oxygen supply in the rat brain. Am J Physiol 228:1896, 1975.
- Perez R, Espinoza M, Riquelme R et al. Arginine vasopressin mediates some of the cardiovascular responses to hypoxemia in fetal sheep. Am J Physiol 256:R1011-R1018, 1989.
- Espinoza M, Riquelme R, Germain AM et al. Role of endogenous opioids in the cardiovascular responses to asphyxia in the fetal sheep. Am J Physiol 256:R1063-R1068, 1989.
- Hohimer AR, Bissonnette JM. Effects of cephalic hypotension, hypertension, and barbiturates on fetal cerebral flow and metabolism. Am J Obstet Gynecol 161:1344, 1989.
- Donegan JH, Traystman RJ, Koehler RC et al. Cerebrovascular hypoxic and auto-regulatory responses during reduced brain metabolism. Am J Physiol 249:H429, 1985.
- Cheek DBC, Hughes SC, Dailey PA et al. Effect of halothane on regional cerebral blood flow and cerebral metabolic oxygen consumption in the fetal lamb in utero. Anesthesiol 67:361-366, 1987.
- Baker BW, Hughes SC, Shnider SM et al. Maternal anesthesia and the stressed fetus: Effects of isoflurane on the asphyxiated fetal lamb. Anesthesiol 72:65, 1990.
- Richardson B, Patrick J, Homan J et al. Cerebral oxidative metabolism in fetal sheep with multiple-dose ethanol infusion. Am J Obstet Gynecol 157:1496, 1987.
- Gleason CA, Hotchkiss KJ. Cerebral responses to acute maternal alcohol intoxication in immature fetal sheep. Pediatr Res 31:645, 1992.
- Fujikawa DG, Dwyer BE, Lake RR, Wasterlain CG. Local cerebral glucose utilization during status epilepticus in newborn primates. Am J Physiol 256:C1160, 1989.
- Harris AP, Kodler AC, Gleason CA et al. Cerebral and peripherial circulating responses to intracranial hypertension in l sheep. Circ Res 64:991, 1989.
Cellular Alterations Associated With Perinatal Asphyxia*
Michael V. Johnston, M.D.
Departments of Neurology and Pediatrics
Johns Hopkins University School of Medicine,
Kennedy Krieger Institute, Baltimore, Maryland
Abstract
Asphyxia triggers a cascade of cellular biochemical events that lead to temporary alterations in cellular function and/or cell death. Tissue hypoxia and ischemia lead to depolarization of neuronal membranes, alteration in cellular ion homeostasis and changes in energy metabolism. The changes are accompanied by enhanced release and diminished reuptake of neurotransmitters, including the excitatory amino acid glutamate. Abnormal accumulation of calcium in neurons is produced by several factors, including opening of voltage-sensitive calcium channels, activation of excitatory amino acid mediated ion channels, diminished pumping of calcium out of neurons, and increased release of free calcium from the endoplasmic reticulum. Elevated intracellular calcium levels appear to kill cells by activation of proteases, lipases, protein kinase C, and generation of free radicals. These factors act synergistically over minutes to hours to produce cellular necrosis. Current research is directed at defining the relative contribution of these steps to cell death and to devising therapeutic strategies to salvage brain tissue.
Introduction
Experimental evidence indicates that asphyxia triggers a cascade of biochemical events that lead to brain dysfunction and eventually to neuronal necrosis (Figure 1). These events are thought to take place over minutes to hours after a threshold of metabolic disruption has been reached.1.,2 Critical levels of ischemia and tissue hypoxia cause tissue to become depolarized and energy metabolism disrupted. In part, these changes appear to be triggered by reduction in delivery of energy substrates such as glucose.3 The combination of inadequate energy substrates and depolarization of neuronal tissue is associated with alterations in synaptic function and accumulation of intracellular calcium levels, two events that appear to be major determinants of neuronal damage. The syndrome of hypoxic-ischemic encephalopathy can be considered as the reflection these pathological alterations in neuronal functions (Table 1). Seizures in the post-asphyxial period probably reflect combinations of altered neuronal ion homeostasis and excessive release of excitatory amino acid neurotransmitters. Coma and other behavioral disturbances probably reflect major disruptions in neuronal metabolism especially in the reticular activating system. Cerebral edema, caused by a breakdown in the brain's ability to regulate cellular water balance, contributes to coma. A detailed understanding of these events can contribute to better ways to salvage brain tissue.
FIGURE 1: Pathophysiology of Hypoxic-Ischemic Encephalopathy
TABLE 1: Clincial Features of Hypoxic-Ischemic Encephalopathy and Potential Cellular Correlates
| CLINICAL FEATURE | POSSIBLE CELLULAR CORRELATE |
|---|---|
| Neonatal Seizures | Overstimulation of excitory amino acid receptors
Membrane depolarization |
| Respiratory depression poor suck |
Disrupted excitory amino acid neurotransmission in brainstem |
| Coma | Metabolic disruption of ascending reticular activating system Disruption of gene expression in immature neurons |
| Brain edema | Disrupted ability to pump water out of neurons; breakdown of blood-brain barrier |
Disruptions in Energy Metabolism
Disruptions in oxidative energy metabolism reflected by reductions in ATP generated from glucose and elevated lactate levels have been a focus of attention as major cellular alterations in asphyxia.4 Although important, the effects of these changes are complex and they may not be primary triggers of damage. The effects of tissue lactate levels in rodent models have been reexamined recently and the results suggest that hydrogen ions associated with lactic acid may be less toxic in the immature rodent brain than in the adult.2,5 In contrast to the adult, where elevated plasma glucose has been associated with increased brain lactic acid and enhanced injury during ischemia, glucose supplementation may reduce injury in certain circumstances.6,7 This may be related to the fact that hypoxic-ischemic brain may contain a reduced concentration of tissue glucose.
Disruptions in oxidative mitochondrial metabolism may also contribute to brain dysfunction and cellular injury from asphyxia.8,9 Damage to mitochondria may occur through 'oxidative stress' in the reperfusion period when free radicals are generated from oxygen.10- 12 Damage to critical enzymes in the respiratory chain, including cytochrome oxidase, occurs following hypoxia-ischemia in animal models.13 Reduced amounts of glutathione, a free radical scavenger, may also contribute to ischemic injury.14 The role of these events in asphyxial damage in the fetus and infant is currently under study.
Disruption of Synaptic Function
Research in animal models over the last decade indicates that the function of certain synapses, especially those that use excitatory amino acids for neurotransmission, is disrupted in brain during hypoxia-ischemia (Figure 2).15-17 Experiments examining the tumover of catecholamines and release of excitatory neurotransmitters into the brain's extracellular fluid using microdialysis show that hypoxia-ischemia causes a marked transient increase in extracellular levels of neurotransmitters.18-20 CSF levels of aspartate have been found to be elevated following asphyxia in human newborns.21 Elevated concentrations of excitatory amino acids in the brain's extracellular space can over-stimulate postsynatic excitatory amino acid receptors linked to ion channels and second messenger systems. Therefore, synaptic dysfunction caused by membrane depolarization and deficits in energy metabolism provide a potentially important link in the chain of events mediating neuronal death.
The elevated levels of extracellular neurotransmitters appear to be caused by a combination of enhanced release and diminished re-uptake into the synaptic nerve terminals.12,15,22 In the unilateral carotid ligation hypoxia model in rodents, a combination of hypoxia and ischemia produces an acute disruption in the ability of synaptosomes to take up glutamate in a sodium dependent manner (Figure 2). This defect can be modified in vitro by adding serum albumin to the incubation medium, suggesting that the effect may be related to accumulation of fatty acids within the cellular membranes. This experiment demonstrates that the defect in glutamate uptake is not related to neuronal death but to an acute and reversible change in glutamate uptake mechanisms. It is noteworthy that injections of a glutamate agonist, N-methyl-D-aspartate, also reproduces this acute change in glutamate uptake into presynaptic nerve terminals.23 This suggests that overstimulation of NMDA-type glutamate receptors may initiate a chain of events which causes a reduction in glutamate uptake, which in turn could trigger a cycle of events that would further increase cellular damage. Pathology of neurotransmitter reuptake systems is a potential target for therapeutic intervention.
Potential Role of Overstimulation of Glutamate Receptors
Several lines of evidence suggest that over-stimulation of glutamate receptors may play a role in the pathogenesis of neuronal injury from hypoxia-ischemia.17 Cell culture experiments indicate that blockade of glutamate receptors markedly increases the tolerance of neurons to severe hypoxia.24 In the brains of animals and humans there is a good general correlation between the distribution of glutamate receptors and the locations of cellular injury from hypoxia-ischemia.16,25 Regions that are especially vulnerable in the immature brain include the basal ganglia, cerebral cortex, hippocampus, and cerebellum. Some of these areas express glutamate receptors only in the perinatal period.26,27 In a rodent model of perinatal hypoxia-ischemia, the NMDA-type glutamate antagonist MK-801 is an effective neuroprotective agent.28,29 These observations suggest that the presence of glutamate receptors on neurons may make them more susceptible to hypoxic injury. It is worthwhile to examine several facets of this hypothesis in some detail.
Audioradiographic studies as well as recent molecular cloning experiments have identified families of related glutamate receptor subtypes (Table 2).30-32 These receptors have been generally classified into NMDA-type glutamate receptors and non-NMDA-type glutamate receptors depending on the angonist analogues that preferentially stimulate them. In turn, the non-NMDA-type glutamate receptors have been divided into metabotropic and ionotropic glutamate receptors. All of these receptor subtypes have been implicated in the processes of hypoxic- ischemic neuronal damage.15,33
FIGURE 2: Schematic of Excitatory Amino Acid Synapse
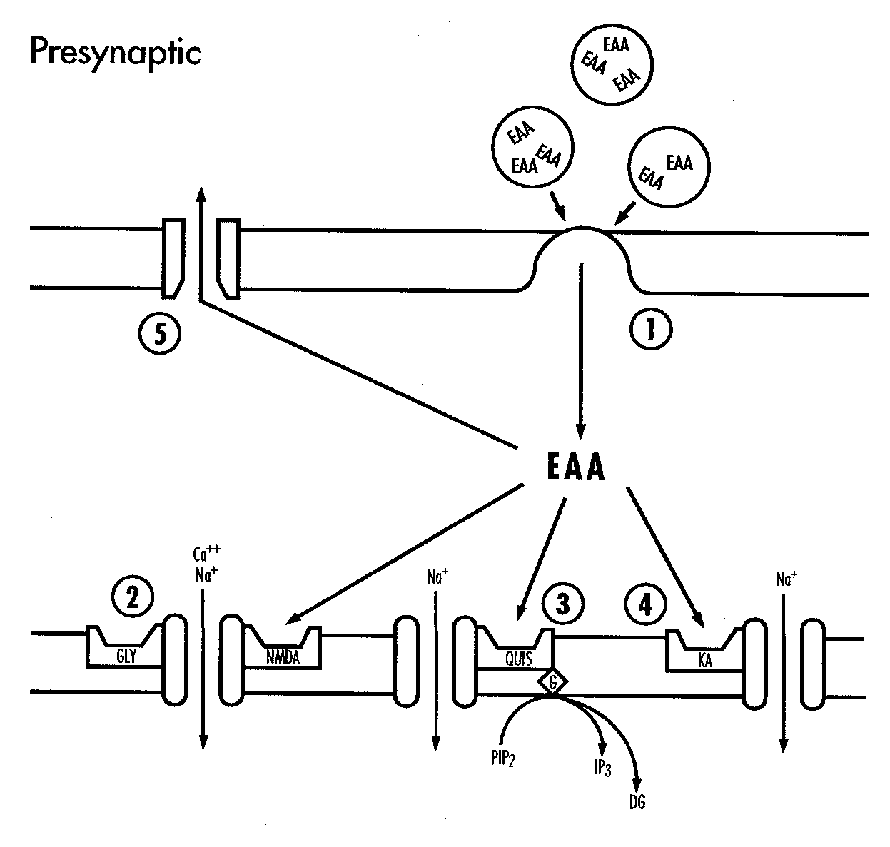
TABLE 2: Classification of Glutamate Receptors
| NDMA-TYPE RECEPTORS |
|---|
| Ionotropic receptor/channel complex |
| NON-NDMA-TYPE RECEPTORS |
|---|
| AMPA ionotropic receptors |
| Kainate ionotropic receptors |
| Metabotropic receptors |
FIGURE 3: Relative Neurotoxicity of Amino Acid Analogues at Different Ages in Rat
FIGURE 4: Ontogeny of Binding to Receptors on the NMDA Receptor Channel Complex in Stratum Oriens (A) and Stratum Radiatum (B) of Rodent Hippocampus
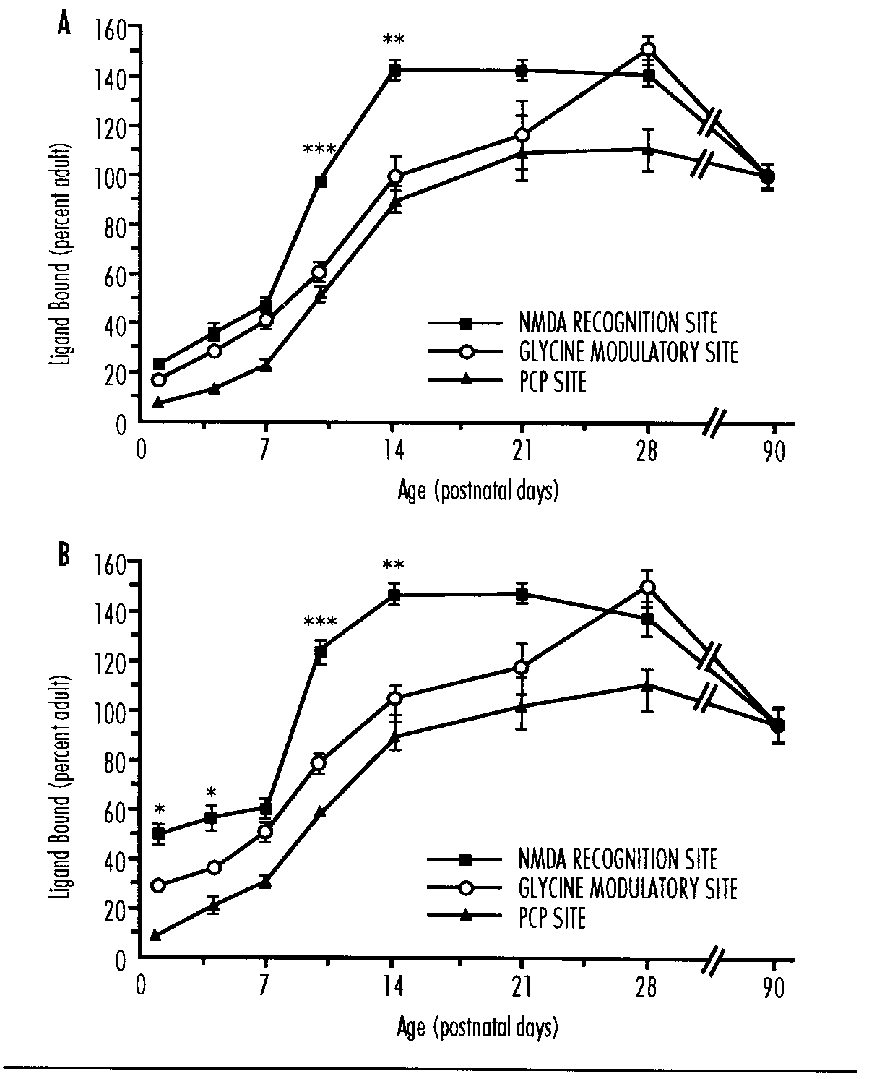
However, different receptor subpopulations appear to be more susceptible to overstimulation at different ages. The neonatal rodent is particularly sensitive to damage from overstimulation of NMDA receptors whereas the adult brain is more sensitive to overstimulation of non- NMDA-type receptors (Figure 3).34 In keeping with this pattern of neurotoxiology, AMPA antagonists appear to be more efficacious than NMDA antagonists against hypoxic-ischemic neuronal injury in adults.33 The histological pattern of injury from NMDA-mediated injury in the neonate is very similar to injury from hypoxia-ischemia.35 The reasons for this age-related change in sensitivity are unclear. NMDA receptors have been shown to be expressed at higher than adult levels in the postnatal period but this overshoot does not occur when the greatest vulnerability to NMDA overstimulation exists (Figure 4).36 This enhanced vulnerability to NMDA overstimulation as well as the efficacy of NMDA-type neuroprotective agents against hypoxia in the neonate, suggests that this type of receptor and channel could play a special role in neonatal asphyxia.
The NMDA receptor channel complex is an interesting macromolecular complex which has recently been cloned (Figure 5).37 The complex includes receptors for glutamate as well as the simple excitatory amino acid, glycine, and a channel which passes both sodium and calcium. A relatively unique feature of this receptor channel complex is that it is ordinarily blocked by the ion magnesium. Blockade of the channel by magnesium is voltage dependent and membrane depolarization is needed to remove this block. This characteristic means that the receptor channel complex can be opened more easily under conditions in which the tissue membranes are depolarized.38 Since this is a frequent occurrence in hypoxic-ischemic tissue39 in asphyxiated infants, NMDA receptor channel complexes would be expected to open more easily under these circumstances (Figure 6). Depolarized, energy deficient neuronal tissue might allow NMDA receptor channel complexes to pass high amounts of calcium even if the extracellular concentration of glutamate is not abnormally elevated. The NMDA receptor channel complex could allow hypoxia-ischemia to sensitize neuronal tissue to modest elevations in extracellular glutamate.
Experimental evidence also implicates non-NMDA glutamate receptors in the pathogenesis of hypoxic-ischemic neuronal injury.33 These receptors are linked to channels that primarily pass sodium into the cell. However, recent neurobiologic evidence indicates that the channels are heteromeric, meaning that they are assembled from several different subunits.31 Developmental studies of gene expression for non-NMDA receptors showed that they are developmentally regulated.40 In the immature brain, non-NMDA-type (AMPA) glutamate receptors appear to have the ability to pass more calcium, as well as sodium, than is passed by adult channels. This unique characteristic of immature non-NMDA receptor channels might allow them to contribute to toxicity from increased concentration of intracellular calcium.
In the immature brain, glutamate receptors appear to play important developmental roles mediating activity dependent plasticity. This refers to changes in brain circuitry mediated by neuronal electrical activity. For example, changes in the organization of ocular dominance columns in visual cortex, based on relative changes in visual stimulation from one or the other eye is an example of this activity. Developmental changes in the expression of NMDA and non-NMDA receptor channel complexes appear to be programmed to serve these functions in the immature brain. Damage from overstimulation of excitatory amino acid receptors may be enhanced at certain times during development because the normal physiologic role of the receptors is also enhanced.15
Enhanced Stimulation of Second Messenger Systems
The metabotropic type glutamate receptor is named because it allows glutamate to induce a metabolic change in the phosphoinositol second messenger system rather than an ion channel opening (Figure 2). Stimulation of the metabotrophic receptor by glutamate causes the breakdown of inositol phosphates into IP3 and diacylglycerol.41
FIGURE 5: Receptor Binding Sites on the NMDA Receptor Channel Complex
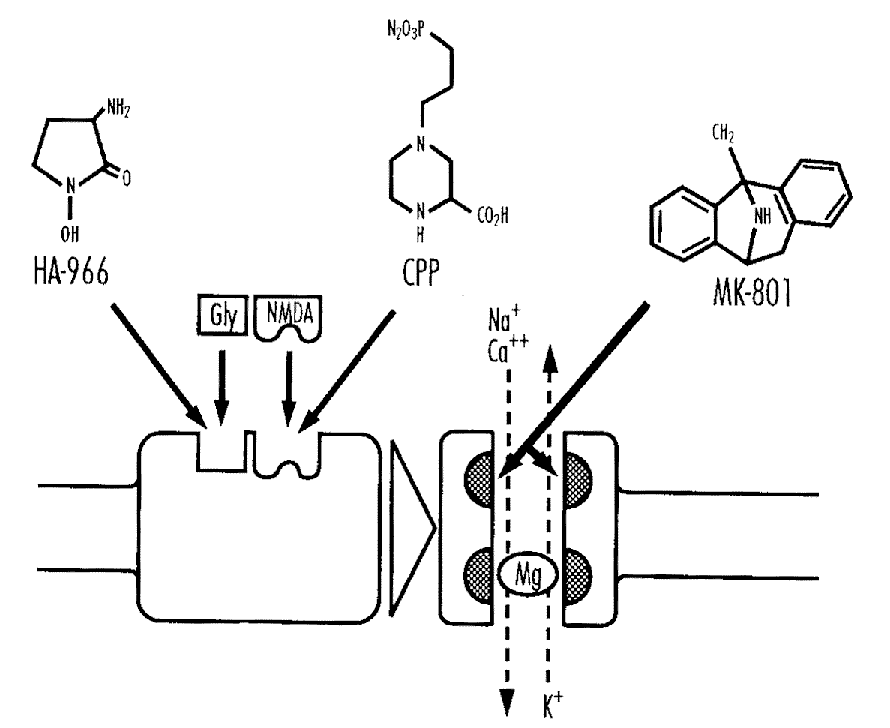
In turn, IP3 stimulates an increase in cytosolic calcium concentrations presumably by releasing calcium from the endoplasmic reticulum. Receptors for the metabotropic glutamate receptor are over-expressed in greater than adult numbers in widespread areas of the rodent and human brain in the perinatal period.26
FIGURE 6: Effect of Depolarization on Opening of the NMDA Receptor Channel Complex
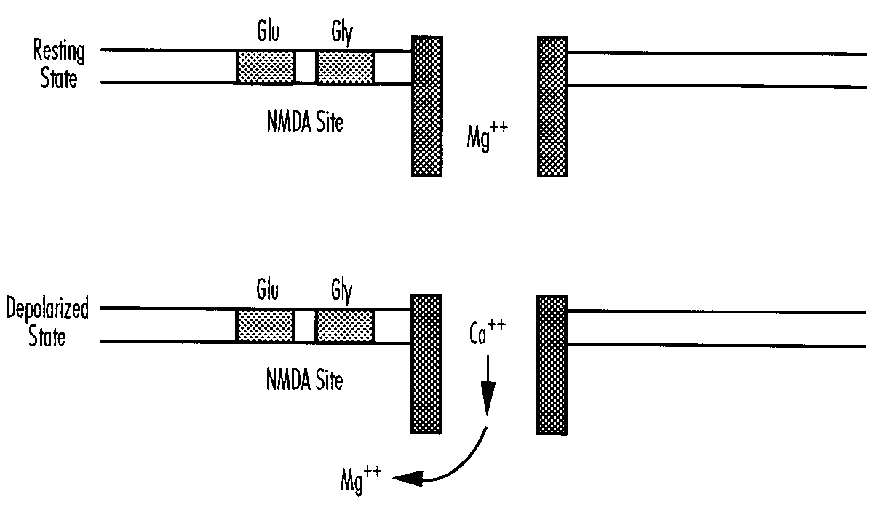
FIGURE 7: Phoshoinositide Turnover Stimulated by the Glutamate Analogue Quisqualate is Enhanced
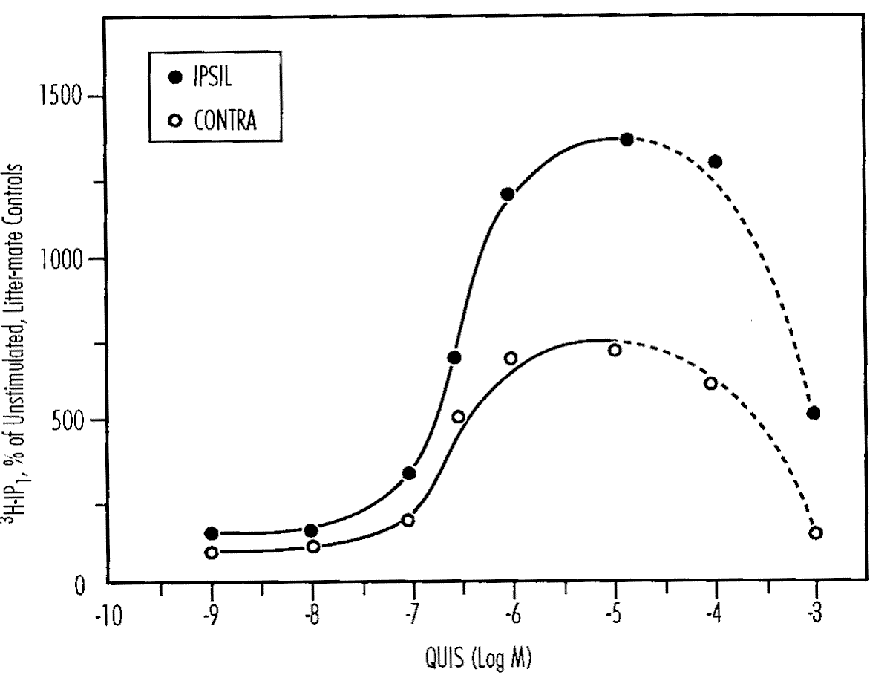
In a rodent model of perinatal hypoxia-ischemia, injury causes an enhancement of the metabotropic stimulated by glutamate response (Figure 7).42 This enhanced response may be related to regenerative changes, since enhanced stimulation for phosphoinositide turnover is already a characteristic of the immature brain. It is noteworthy that this stimulation is specific for glutamate, but not cholinergically- stimulated PI turnover. This interesting phenomenon indicates a specific effect of hypoxia-ischemia on the glutamate synapses.
Changes in Intracellular Calcium Concentrations
The best current theory of neuronal injury from perinatal asphyxia gives a central position to intracellular concentrations of calcium (Figure 8).43-45 Calcium's important regulatory functions, its
FIGURE 8: Schematic of Mechanisms Controlling Calcium Homeostasis in Neurons
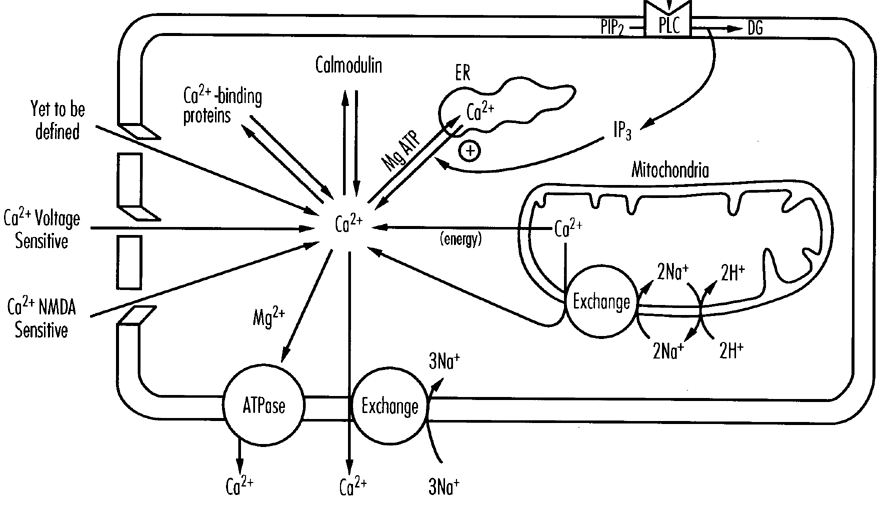
its ability to stimulate a variety of autolytic enzymes, as well as the tight controls that are normally exercised over calcium suggest that it is a potentially destructive molecule.43 Calcium fluxes come under the control of a host of cellular functions including excitatory amino acid receptor channel complexes, ion fluxes across neuronal membranes, the inositol phosphate second messenger system, the endoplasmic reticulum as well as the mitochondrion. The regulation of calcium is particularly likely to be disruptive during asphyxia as numerous events conspire to raise intracellular level concentrations of calcium.1 As membrane ionic gradients drop, both voltage sensitive calcium channels and the voltage sensitive NMDA channel pass more calcium into the cell. Calcium is less adequately extruded in a hypoxic environment because of the reduced efficiency of energy requiring countercurrent pumps. Tissue depolarization also contributes to release of glutamate from presynaptic nerve terminals and this stimulates calcium to pass through AMPA and NMDA receptor channel complexes. Energy deficiency may also impair the calcium-buffering capacity of mitochondria. The summed effect is an enhanced entry of calcium into neurons that are less equipped to reduce this level.
The relative importance of these different calcium-mediated events is still being examined.45 Enhanced calcium entry may enhance metabolic stress on damaged neurons. Calcium activates proteases and lipases that destroy cellular integrity (Figure 9). Calcium also contributes to the production of free radicals such as O2- and OH- produced from the breakdown of lipid membranes, the disruption of mitochondrial respiration and the conversion of hypoxanthine to uric acid.2 These free radicals are particularly likely to attack cellular membranes. A newly recognized free radical, nitric oxide, is produced by conversion of arginine to citrulline in a small number of brain neurons (Figure 10).46 The conversion by nitric oxide synthase is stimulated by calcium. Nitric oxide is thought to have a dual role, as a neurotoxin for adjacent neurons and as a catalyst of cerebral vascular relaxation and vasodilatation.
FIGURE 9: Shematic of Mechanisms of Excitatory Amino Acid Mediated Neurotoxicity
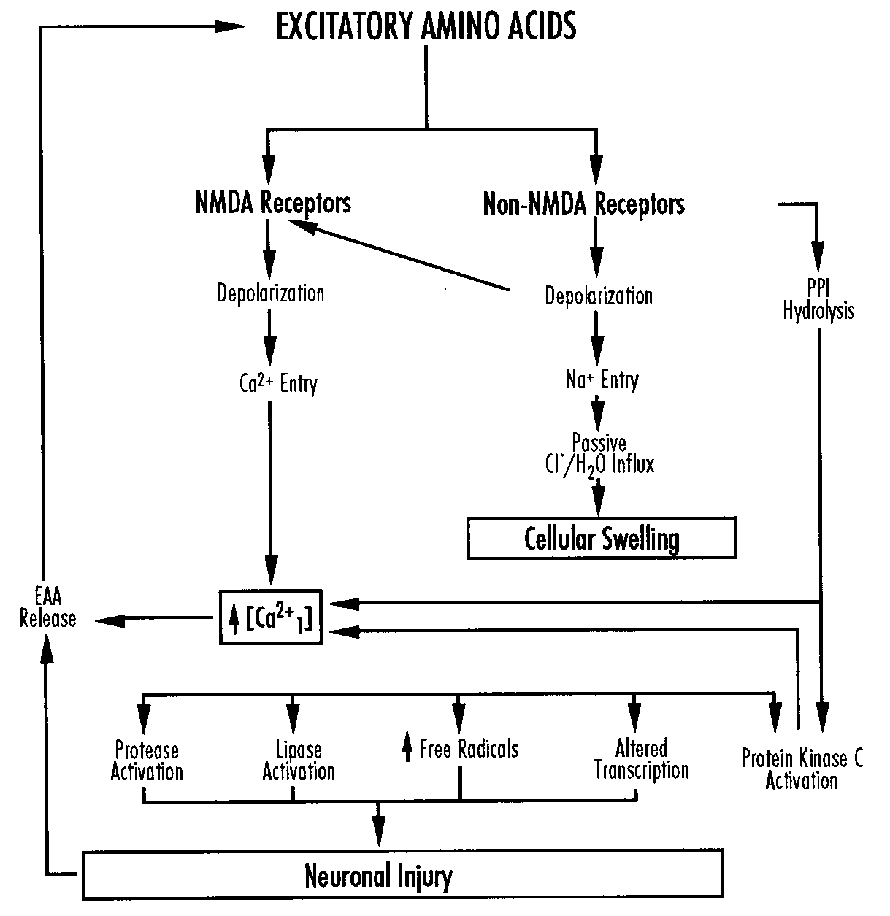
It has been proposed that nitric oxide or a closely related compound is the link between neuronal activity and vasomotor activity. Generation of free radicals appears to be an important transient event in the cellular reactions following asphyxia, and its relevance to therapy is being explored.
Implications for Management
Current management of hypoxic-ischemic encephalopathy is directed at maintenance of adequate blood pressure to support cerebral blood flow, maintenance of normal plasma glucose concentrations, and suppression of seizures.47 The appropriate level of glucose remains controversial in view of experimental work suggesting that higher levels might be helpful.7 The impact of seizures in the context of hypoxia-ischemia is also not known. However, according to the conceptual framework outlined here, seizures might be expected to worsen injury by causing tissue depolarization, neurotransmitter release, and calcium overload. Therefore, vigorous antiseizure therapy is generally prescribed. In the neonate, cerebral edema that occurs with severe hypoxic-ischemic encephalopathy appears to be a sign of injury rather than a contributor to it.2 There is little evidence that therapy directed specifically towards cerebral edema reduces brain damage although prevention of massive edema could prevent lethal herniation.
FIGURE 10: Mechanism of Activation of Nitric Oxide Synthase by Excitatory Amino Acid Neurotransmitters to Produce Nitric Oxide
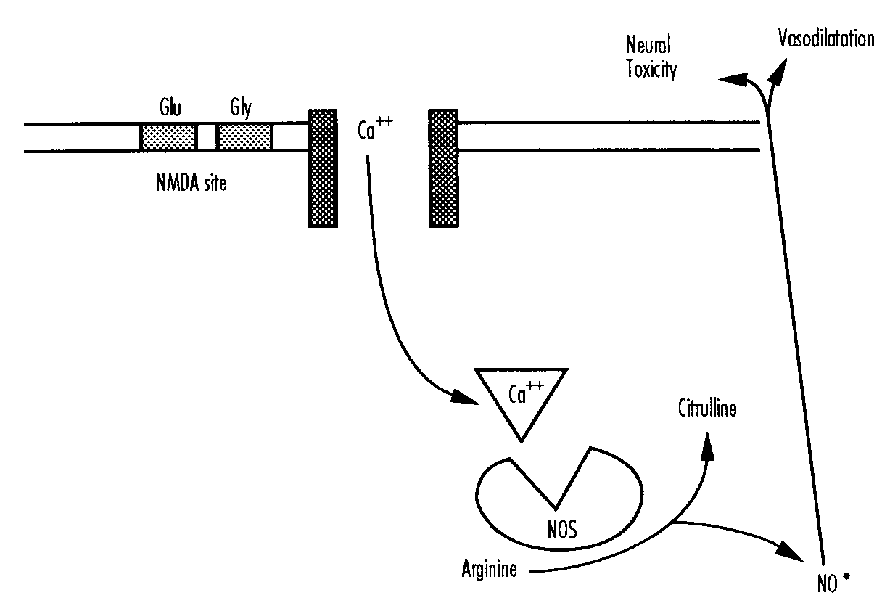
Future therapy to salvage brain during hypoxic-ischemic encephalopathy will be based on an understanding of the cascade of events already discussed and possibly others not identified. It would be worthwhile to distinguish between those that are of primary importance and others that are epiphenomenon. The conceptual framework outlined in this presentation suggests that a number of pharmacologic interventions such as glutamate antagonists, calcium antagonists, drugs that alter intracellular calcium concentrations and others that improve cellular energy metabolism might have a beneficial effect singly or in combination. A clinically approved glutamate antagonist with some therapeutic potential, dextromethorphan, is moderately protective against hypoxic-ischemic damage in animal models.48 We recently used this drug to treat an infant with nonketotic hyperglycinemia, which may damage the brain in part through overstimulation of NMDA receptors.49 This drug and others may hold potential for neuroprotection of the neonatal brain.
Conclusion
Over the last decade a great deal has been learned about the cascade of cellular events in the brain that are triggered by asphyxia. Current research focuses on events triggered by tissue depolarization, overstimulation of excitatory amino acid receptors, and the effects of calcium overload in damaged neurons. These pathological effects may be enhanced in the immature brain because their physiological role is relatively greater at this age than in the adult. The conceptual framework developed through this research is useful for understanding clinical manifestations of asphyxia and developing rational therapy.
References
- Siesjo BK. Cell damage in the brain: A speculative synthesis. J Cereb Blood Flow Metabol 1:155-185, 1981.
- Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: Relation to perinatal brain damage. Pediatr Res 27:317-326, 1990.
- Vannucci RC, Christensen MA, Stein DT. Regional cerebral glucose utilization in the immature rat: Effect of hypoxia-ischemia. Pediatr Res 26:208-214, 1989.
- Myers RE. A unitary theory of causation of anoxic and hypoxic brain pathology. In: Fahn S, David J, Rowland L (eds). Cerebral Hypoxia and Its Consequences. New York: Raven Press, pp. 195-213, 1979.
- Voorhies TM, Rawlinson D, Vannucci RC. Glucose and perinatal hypoxic-ischemic brain damage in the rat. Neurol 36:1115-1118, 1986.
- Pulsinelli WA, Waldman S, Rowlinson D, Plum R. Moderate hyperglycemia augments ischemic brain damage: A neuropathological study in the rat. Neurol 32:1239-1246, 1982.
- Hattori H, Wasterlain CG. Posthypoxic glucose supplement reduces hypoxic-ischemic brain damage in the neonatal rat. Ann Neurol 28:122-128, 1990.
- Brown AW, Brierley JB. The earliest alterations in rat neurons and astrocytes after
anoxia-ischemia. Acta Neuropathol 23:9-22, 1973. - Sims NR, Pulsinelli WA. Altered mitochondrial respiration in selectively vulnerable subregions following transient forebrain ischemia in the rat. J Neurochem 49: 1367-1374, 1987.
- Amstead WM, Mirro R, Busija DW, Leffler CW. Postischemic degeneration of superoxide anion by newborn pig brain. Am J Physiol 255:401-403, 1988.
- Rosenberg AA, Murdaugh E, White CW. The role of oxygen free radicals in post-asphyxia cerebral hypoperfusion in newborn lambs. Pediatr Res 26:215-219, 1989.
- Pellegrini-Giampietro DE, Cherici G, Alesiani M et al. Excitatory amino acid release and free radical formation may tooperate in the genesis of ischemia-induced neuronal damage. J Neurosci 10:1035-1041, 1990.
- Nelson C, Silverstein FS. Acute regionally specific reductions in cytochrome oxidase activity predict selective vulnerability in perinatal rodent brain. Ann Neurol 30:482, 1991.
- Mizui T, Kinouchi H, Chan PH. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am J Physiol 262:H313-H317, 1992.
- McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41-70, 1990.
- Choi AW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1:623-634, 1988.
- Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol 19:105-111, 1986.
- Silverstein FS, Johnson MV. Effects of hypoxia-ischemia on monoamine metabolism in the immature brain. Ann Neurol 15:342-347, 1984.
- Benveniste HJ, Drejer A, Schousboe A, Diemer NH. Elevation of the extracellular
concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43:1369-1374, 1984. - Silverstein FS, Naik B, Simpson J. Hypoxia-ischemia stimulates hippocampal glutamate efflux in perinatal rat brain: An in vivo microdialysis study. Pediatr Res 20:587-590, 1991.
- Rikonen RS, Kero PO, Simell OG. Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr Neurol 8:37-40, 1992.
- Silverstein FS, Buchanan K, Johnston MV. Perinatal hypoxic-ischemia disrupts striatal high affinity 3H-glutamate uptake into synaptosomes. J Neurochem 47:1614-1619, 1986.
- Hu B, McDonald JW, Johnston MV, Silverstein FS. Excitotoxic brain injury suppresses striatal high affinity glutamate uptake in perinatal rats. J Neurochem 56:933-937, 1991.
- Rothman S. Synaptic release of excitatory amino acid neurotransmitters mediates anoxic neuronal death. J Neurosci 4:1884-1891, 1984.
- Silverstein FS, Torke L, Barks J, Johnston MV. Hypoxia-ischemia produces focal disruption of glutamate receptors in developing brain. Brain Res 34:33-39, 1987.
- Greenamyre JT, Penney JB, Young AB et al. Evidence for a transient perinatal glutamatergic innervation of globus pallidus. J Neurosci 7:1022 -1030, 1987.
- Barks JDE, Silverstein FS, Sims K et al. Glutamate recognition sites in human fetal brain. Neurosci Lett 84:131-136, 1988.
- McDonald JW, Silverstein FS, Johnston MV. MK-801 protects the neonatal brain from hypoxic-ischemic injury. Euro J Pharmacol 140:359-361, 1987.
- Hattori H, Main AM, Schwartz PH et al. Posthypoxic treatment with MK-801 reduces hypoxic-ischemic damage in the neonatal rat. Neurol 39:713- 718, 1989.
- Young A, Fagg GE. Excitatory amino acid receptors in the brain: Membrane binding and receptor autoradiographic approaches. Trends Pharm Sci 11:126-133, 1990.
- Gasic GR, Hollman M. Molecular neurobiology of glutamate receptors. Ann Rev Physiol 54:507-536, 1992.
- Henneberry RC. Cloning of the genes for excitatory amino acid receptors. Bio Essays 14:332-338, 1992.
- Buchan AM, Xue D, Huang Z-G et al. Delayed AMPA receptor blockade reduces cerebral infarction induced by focal ischemia. Neuro Report 2:473- 476, 1991.
- McDonald JW, Silverstein FS, Johnston MV. Neurotoxicity of N-methyl-D-aspartate is markedly enhanced in the developing rat central nervous system. Brain Res 459:200-203, 1988.
- Ikonomidou C, Mosinger JL, Shahid Salles K et al. Sensitivity of the developing rat brain to hypobaric/ischemic damage parallels sensitivity to N-methyl-D-aspartate neurotoxicity. J Neurosci 9:2809-2818, 1989.
- McDonald JW, Johnston MV, Young AB. Differential ontogenic development of three receptors comprising the NMDA receptor/channel complex in the rat hippocampus. Exp Neurol 110:237-247, 1990.
- Monyer H, Sprengel R, Schoepfer R et al. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science 256:1217-1221, 1992.
- Henneberry RC, Novelli A, Cox JA, Lysko PG. Neurotoxicity at the N-methyl-D-aspartate receptor in energy compromised neurons—a hypothesis for cell death in aging and disease. Ann NY Acad Sci 568:225-233, 1989.
- Vornov JJ, Coyle JT. Enhancement of NMDA receptor-mediated neurotoxicity in the hippocampal slice by depolarization and ischemia. Brain Res 555:99-106, 1991.
- Pelligrini-Giampietro DE, Bennett MVL, Zukin RS. Differential expression of three glutamate receptor genes in developing rat brain: An in situ hybridization study. Proc Natl Acad Sci 88:4157-4161, 1991.
- Nicoletti F, Iadorola MJ, Curobleiuski JT, Costa E. Excitatory amino acid recognition sites coupled with inositol phospholipid metabolism: Developmental changes and interaction with adrenoreceptors. Proc Natl Acad Sci 83:1931-1935, 1986.
- Chen C, Silverstein FS, Fisher SK et al. Perinatal hypoxic-ischemic brain injury enhances quisqualic acid stimulated phosphoinositide turnover. J Neuroc hem 51:353-359, 1988.
- Choi DW. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci 11:465-469, 1988.
- Johnston MV, Silverstein FS. New insights into mechanisms of neuronal damage in the developing brain. Pediatr Neurosci 12:87-89, 1986.
- Pulsinelli WA, Jacewicz M, Buchan AM. Stroke and hypoxic-ischemic disorders. In: Johnston MV, MacDonald RL, Young AB (eds). Principles of Drug Therapy in Neurology. Philadelphia: F.A. Davis, pp.118-160, 1992.
- Bredt DS, Hwang PM, Synder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 347:768-770, 1989.
- Johnston MV, Donn S. Hypoxic-ischemic encephalopathy In: Nelson N (ed). Current Therapy in Neonatal Perinatal Medicine. Philadelphia: B.C. Decker, Inc., pp. 276-279, 1990.
- Steinberg GK, Kunis D, Slaeh J, de la Paz R. Protection after transient focal cerebral ischemia by the N-methyl-D-aspartate antagonist dextromethorphan is dependent upon plasma and brain levels. J Cereb Blood Flow Metab 11:1015-1024, 1991.
- Hamosh A, McDonald JW, Valle D et al. Experience with dextromethorphan and high dose benzoate therapy in an infant with non-ketotic hyperglycinemia. J Pediatr 121:131-135, 1992.
Hypoxia and Cerebral Blood Flow in the Human
Gorm Greisen, M.D.
Department of Neonatology
Rigshospitalet, Copenhagen, Denmark
Introduction
Nothing is known of the acute effects of hypoxia on cerebral blood flow (CBF) in the human fetus and virtually nothing is known in the human neonate. The basis for discussing the effects of pure hypoxia as separate from the effects of the complex patterns of asphyxia is minimal. In this paper, data from the human—mostly preterm—neonate is presented in the light of evidence from the adult human, and perinatal animal.
Hypoxemia and Global CBF
One of the first patients I studied by the intravenous 133-Xe clearance method was a preterm infant on CPAP. We did simultaneous recording of Doppler flow velocity and when we recorded a very high flow velocity in the internal carotid artery we realized that he was apneic and deeply cyanotic. This was 3 minutes after tracer injection. He quickly responded to tactile stimulation. The resulting global CBF value was 57.6 m1/100g/min. I never used the result, and it was only later I realized that the value was three times higher than the value measured on the next day.
Already in 1948 Kety and Schmidt1 had demonstrated that hypoxemia increases CBF in healthy adults, and many more studies on this followed, mainly motivated by aviation medicine. It was not until 1991, however, that the effects of hypoxia was studied systematically in newborn infants.2 Unfortunately, the technique used was near-infrared spectrophotometry which allows continuous monitoring of cerebral hemoglobin content, and hence of cerebral blood volume, but which only indirectly indicate changes in CBF. Spontaneous as well as induced decreases in arterial oxygen saturation resulted in a gradual cerebral hemoglobin desaturation, and a small increase in total cerebral hemoglobin concentration, both of which returned to baseline when the hypoxemia was relieved. A similar reaction has been reported during routine endotrachael suctioning,3 which produced a consistent drop in arterial oxygen saturation. Endotracheal suctioning obviously produces a variety of physiological effects,4 but this particular response could be much reduced by preoxygenation. Although the evidence thus is far from complete, it is reasonable to assume that CBF in newborn—and even preterm infants—increases promptly—within seconds—in response to hypoxemia, as it does in adult man and in perinatal animals.
Hypoxemic Thresholds
At what level of hypoxemia does CBF increase and how much? These questions can only at the present time, be answered indirectly. The relation between oxygen tension and hemoglobin saturation has the well known sigmodeal shape, the location of the dissociation curve however depends on a number of factors, the most important being blood pH, DPG concentration, CO2 tension, and the fraction of fetal hemoglobin. The half- saturation point may vary from 2.5 kPa (20 torr) to 4 kPa (30 torr). With a half-saturation point—P50—in the low range, noticeable desaturation will only occur at oxygen tensions of 7.5 kPa (55 torr) or below. It is therefore not surprising that only little increase of CBF was seen in adult dogs ventilated with 10 percent oxygen, whereas marked increase occurred with 6 percent,5 pointing to a 'threshold' at 50-60 torr (7-8 kPa). At oxygen tensions below this level—with desaturation—cerebral oxygen delivery is maintained by up to twofold to threefold increases in CBF. The relation between arterial oxygen saturation and CBF is not a proper hyperbola, since oxygen extraction increases in parallel with CBF.
Oxygen is used at the mitochondrial level, and oxygen transport depends on diffusion as well as oxygen delivery. The critical quantity is the oxygen tension in the venous end of the capillary, since this represents the diffusion driving pressure to the mitochondria of the most depriviledged neurons. When the oxygen supply to the mitochondria fails, neurons will shut off their synaptic activity. Brief hypoxemia switches off the spontaneous EEG activity in preterm infants.6 Visual evoked responses in preterm infants were also consistently affected at arterial oxygen tensions of 2.5 kPa.7 A theoretic calculation from this data supports that the oxygen extraction fraction cannot get below 0.1-0.15, corresponding to a maximal hypoxemia-induced twofold to threefold increase of CBF even in preterm infants.
Hypoxemia and Distribution of CBF
Thus far, only global—average—CBF has been discussed. Obviously, the brain is inhomogeously perfused—and brain damage due to hypoxic-ischemic injury is not uniform, but rather tend to have a characteristic distribution. It is therefore natural to suppose that hypoxemia, in some way may change the distribution of CBF. There is good evidence that this is so in perinatal animals. The first evidence was published by Professor Kjellmer and his coworkers in 1978,8 who noted that hypoxic gas mixture to the ewe increased rCBF more in the brainstem of the chronically-instrumented fetal lamb than in its hemispheres.
This has been reproduced in the newborn puppy, which is more immature at birth than the term fetal lamb. During extreme hypoxemia— PaO2=11 torr/1.5 kPa—flow to the brainstem increased threefold to fivefold,9 without increases in local glycolysis, whereas in the hemispheres, and in white matter in particular, the blood flow increase was moderate at 1.5-2 fold, resulting in marked anaerobic glycolysis. There is evidence that this redistribution is mediated through the sympathic system, since it may be modified by alpha-blockade in the puppy10 or sympathectomy in the piglet.11
There is no published data in the human, but preliminary results in Copenhagen obtained by single-photon emission tomography using 99m-Tc- labelled HMPAO has shown that the high basal flow to the central parts of the brain was relatively reduced rather than increased during hypoxemia.12
Delayed Hyperperfusion
How does CBF react beyond the immediate period following hypoxemia? Little is known of this from experiments on animals, mainly because of the difficulty of longterm intensive care of animals with multisystem hypoxic injury. But in adult humans, a late stage of cerebral hyperperfusion has been demonstrated following cardiac arrest.13 The hyperperfusion peaked after about 24 hours, was associated with severe EEG depression, and carried a grave prognosis. A very similar picture has been described in the newborn. A small group of severely asphyxiated infants was characterized by electrocerebral silence, twofold to threefold increased CBF, loss of normal CBF reactivity, and subsequent evidence of global severe brain injury.14 Cerebral blood flow velocity may increase late, after the first days of life,15 and in a few severely asphyxiated infants internal jugular vein blood sampling has showed that the hyperperfusion is not associated with an elevated CMRO 2,16 classifying the state as one of luxury perfusion. In a single severely asphyxiated infant a combination of cerebroelectrical silence, low global CBF and high cerebrovenous saturation was associated with severe brain injury, apparently occurring several weeks before birth.17 This suggests that the measurement of CBF may be useful for diagnostic and/or prognostic purposes.
To conclude, CBF may increase twofold to threefold in acute hypoxemia, provided perfusion pressure is maintained. This, combined with increased oxygen extraction, will allow electrical function to persist until arterial oxygen saturation falls below 50 percent. In asphyxia the situation is more complex, and a 'redistribution' within the brain has been consistently demonstrated in perinatal animals, mediated through the sympatoadrenergic system. This may, however, not be so in the human. In human adults, and human newborns, a state of delayed luxury perfusion with lost reactivity has been well documented which carries a grave prognosis. It is, however, unclear if this cerebrovascular disturbance represents a cause of neuronal injury, or merely a result.
References
- Kety SS, Schmidt CF. The effects of altered arterial tensions of carbon dioxide and oxygen on cerebral blood flow and cerebral oxygen consumption of normal young men. J Clin Invest 27:484, 1948.
- Livera LN, Spencer SA, Thorneiley MS et al. Effects of hypoxemia and bradycardia on neonatal cerebral haemodynamics. Arch Dis Child 66:376, 1991.
- Shah AR, Kurth CD, Gwiazdowski SG et al. Fluctuations in cerebral oxygenation and blood volume during endotracheal suctioning in premature infants. J Pediatr 120:769, 1992.
- Skov L, Ryding J, Pryds O, Greisen G. Changes in cerebral oxygenation and cerebral blood volume during endotracheal suctioning in ventilated neonates. Acta Paediatr Scand 81:389, 1992.
- Kogure K, Scheinberg P, Reinmuth OM et al. Mechanisms of cerebral vasodilatation in hypoxia. J Appl Physiol 29:223, 1970.
- Roberton NRC. Effect of acute hypoxia on blood pressure and electroencephalogram of newborn babies. Arch Dis Child 44:719, 1969.
- Pryds O, Greisen G. Preservation of single-flash visual evoked potentials at very low cerebral oxygen delivery in preterm infants. Pediatr Neurol 6:151, 1990.
- Blomstrand S, Karlson K, Kjellmer I. Measurement of cerebral blood flow in the fetal lamb with a note on the flow distribution. Acta Physiol Scand 103:1, 1978.
- Cavazutti M, Duffy TE. Regulation of local cerebral blood flow in normal and hypoxic newborn dogs. Ann Neurol 11:247, 1982.
- Hernandez MJ, Hawkins RA, Brennan RW. Sympathetic control of regional cerebral blood flow in the asphyxiated newborn dog. In: Heistad DD, Marcus ML (eds). Cerebral Blood Flow, Effects of Nerves and Neurotransmitters. New York: Elsevier, pp. 359, 1982.
- Goplerud JM, Wagerle LC, Delivoria-Papadopoulos M. Sympathetic nerve modulation of regional cerebral blood flow during asphyxia in newborn piglets. Am J Physiol 60:H1575, 1991.
- Börch K, Greisen G. rCBF in hypoxemic or hypotensive preterm infants. Preliminary results. Nordic CBF workshop. Hilleröd, Abstract #2.6, 1993.
- Cohan SL, Seong KM, Petite J et al. Cerebral blood flow in humans following resuscitation from cardiac arrest. Stroke 20:761, 1989.
- Pryds O, Greisen G, Lou H et al. Vasoparalysis associated with brain damage in asphyxiated term infants. J Pediatr 117:119, 1990.
- Levene MI, Fenton AC, Evans DH et al. Severe birth asphyxia and abnormal cerebral blood flow velocity. Dev Med Child Neurol 31:427, 1989.
- Frewen TC, Kissoon N, Kronick J et a1. Cerebral blood flow, cross-brain oxygen extraction, and fontanelle pressure after hypoxic-ischemic injury in newborn infants. J Pediatr 118:265, 1991.
- Skov L, Pryds O, Greisen G, Lou H. Estimation of cerebral venous saturation in newborn infants by near infrared spectroscopy. Pediatr Res 33:52, 1993.
Hypoxia Opportunism During Brain Development*
Philippe Evrard, M.D., Jean-François
Gadisseux, M.D., and Pierre Gressens, M.D.
Service de Neurologie Pédiatrique, Hôpital
Universitaire Saint-Luc and Laboratoire de
Neurologie du Développement, University
of Louvain Medical School at Brussels, Belgium
Introduction
Neuronal and glial hypoxia/ischemia are unfavorable environmental determinants of central nervous system (CNS) development. At most neurodevelopmental steps, the hypoxia opportunism exploits the developmental mechanisms to produce abnormal morphogenetic patterns and functional impairments. The aim of this paper is to review a few conceptual and methodological tools permitting analysis of the influence of hypoxic and other environmental determinants on developing neural tissue during embryonic and fetal life1 (Figure 1).
Preparation of the Neural Germinative Epithelium
In a previous paper,2 we proposed to call "period of preparation of the neural germinative epithelium" (or phase II of neurogenesis) the developmental period between the isolation of the neural groove and the onset of neuronal migration. In order to assess the environmental influences acting during this neurodevelopmental phase, we used whole post-implantation mouse embryo cultures from 5 to 26 somites,2 in 80 percent human serum and 20 percent rat serum which provide an optimal nutritional environment. The optimal gas phase is 5 percent CO2 and 95 percent air. The gas, sera, and other medium parameters provide the opportunity to study the influences of nutrients and of CO2-O2 concentrations on brain growth at these early periods of brain development. The embryos, including their yolk-sac (vitelline) circulation and their heart beats, are examined under a stereomicroscope. Histological examination includes morphometric evaluation and immunocytological methods (RC2, vimentin and GFAP). Both scanning and transmission electron microscopic examinations are performed. At the ultrastructural level, the glial lineage is studied with Gadisseux and Evrard's method based upon the preservation and staining of particulate glycogen.3,4 Applied to this model, cocaine severely interferes with the differentiation of radial glial cells (RGCs), perhaps through the activation of intermediate early genes such as c-fos. The subsequent neocortical pattern reflects this severe defect of RGCs.5 We had not previously confirmed cocaine-induced reduction of vitelline artery diameter in our model.6Ethanol dramatically enhances cell death in the ventromedial part of the mesencephalic-prosencephalic junction of the primitive neuroepithelium.7Retinoic acid and valproate were also applied on these explants in our laboratory. They provoked developmental lesions of the neuroepithelium [see reference 2 for retinoic acid; the results concerning valproate are in press]; retinoic acid and valproate do not seem to influence the vitelline circulation. The application of vasoactive intestinal peptide (VIP) to these whole cultured mouse embryos suggest that VIP seems to be a crucial determinant of embryonic growth, including CNS growth;8 in vivo, extraembryonic VIP could be the source of this regulating factor of embryonic and CNS growth.8 The observations stress the importance of maternal and/or placental environment and the crucial influence of the circulatory factors.
FIGURE 1: Schematic Representation of the Chronology of the Main Development Events
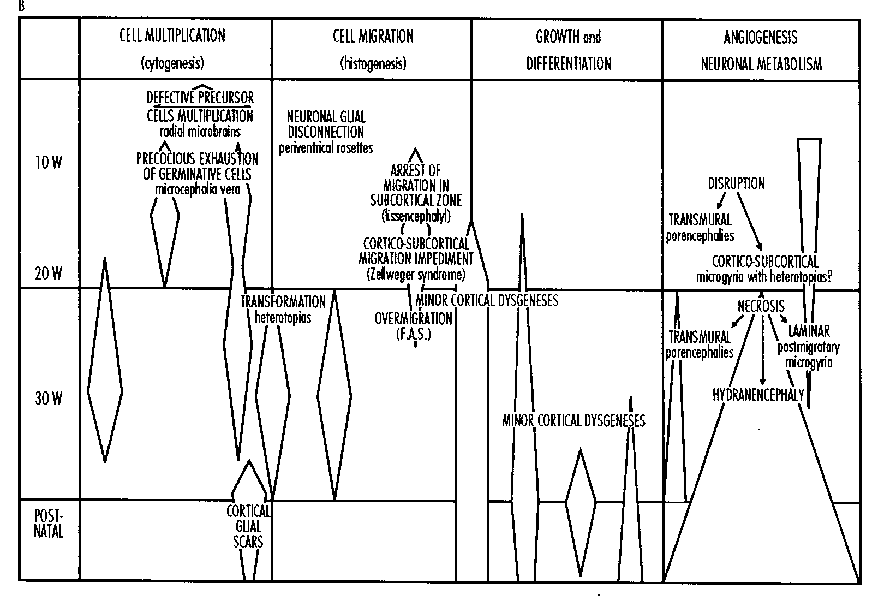
Energy Supply for Migrating Neurons
Migrating neurons, often distant from blood vessels, seem to use primarily anaerobic glycolysis*.They are in close contact with RGCs, which are stuffed with particulate glycogen,9 when all normal neuronal elements are devoid of any detectable particulate glycogen.3 RGCs extend from the ventricular zone (close to the choroid plexuses and vessels) to the pial surface, an area richly supplied by the leptomeningeal plexus. The RGCs, fully and uniformly loaded with particulate glycogen, seem to act as energy corridors. We have no information concerning the mechanism of glucose transfer from glial glycogen to the migrating neuron. We ignore also the destiny of the abundant lactate, probably produced anaerobically by migrating neurons. The developing neurons transform from a predominantly anaerobic metabolism when migrating towards a more aerobic condition, after their postmigratory settlement in a more highly vascularized cortical plate. This neuronal metabolic transformation could be approached by the morphological study of the mitochondrial volume which could triple in aerobiosis. Angiogenests parallels this metabolic transformation and is summarized in Figure 1, including the data recently published by Kuban and Gilles10 and Norman and O'Kusky.11
Outside the general metabolic schedule we just summarized, to our knowledge, no fine timing with detailed precision about regional variation is available concerning the developmental transformation of neuronal metabolism from anaerobiosis into aerobiosis. Among the sparse data, we know that neocortical infragranular neurons become oxygen dependent at mid gestation (see the next paragraph, concerning the laminar necrosis). Synaptogenesis carries other, more hypothetical, information concerning metabolism: the modest efficiency of anaerobic metabolism could probably not cover the endothermic character of intense synaptogenesis. This limiting factor for synaptogenesis is quite consistent with the data summarized in Figure 1.
The Postmigratory Infragranular Layers: A Sensitive Target for Perfusion Failures/Hypoxia During the Second Half of the Middle Trimester of Gestation
Since the 1974 seminal contribution from Caviness and coworkers on the etiology of brain malformations,12 the basic lesion in a significant number of cases of polymicrogyria has been interpreted as a postmigratory laminar necrosis predominating in the infragranular part of the cortical plate and especially in the neocortical layer V.13 Clinicopathologic correlations, including sequential ultrasound, from the European Multicenter Study,14 confirm with certainty that perfusion failures leading to layered polymicrogyrias happened in the collected cases after the end of neuronal migration and before the peak of gyration (between 20 and 30 weeks of gestation). Using local injections of the excitotoxin ibotenic acid (ibo), Innocenti and Berbel15 created a kitten model of neocortical destruction, followed by a subsequent renewal of neuronal migration if the ibo injection is not performed too late and does not destroy the whole neuronal complement. Ibotenic acid mimics the hypoxic insult remarkably well, i.e., it kills postmigration neurons; the striking feature is that migrating neurons are almost fully preserved and devoid of any sign of toxicity due to ibotenate. We have produced very precise laminar necrosis mimicking human microgyria and some human neocortical dysplasia using ibo injections in the neocortex of newborn mice. Other variants of microgyrias, which occur before 20 weeks of gestation, are represented in several series in the literature.13,14,16 The comparative chronology of angiogenesis, neuronal metabolism, and neuronal migration (Figure 1) provides a framework for the interpretation of the pathophysiology of the different types of microgyria. The fact that, after hypoxic insults, neuronal displacements along or outside of the migratory corridors can occur after the end of normal neuronal migration has also to be taken into account when comparing the variants of microgyria. During almost two decades, developmental pathologists have entertained controversial and opposite beliefs about microgyrias. The progress of our knowledge about brain development and hypoxia may provide adequate tools to reconcile the protagonists on the basis of newly recognized developmental mechanisms. The precise timing of events during the weeks and months before and after 20 weeks of gestation, the slight individual dyschronologies between the different developmental aspects summarized in the adjacent columns of Table 1, and the extreme variety of severity and topography of the causal agents reveal dozens of patterns and subpatterns that may be produced and explain rather easily most variants of microgyrias.
TABLE 1: Hypoxia, Ischemia, Perfusion Failures: A Few Types and Pathophysiological Mechanisms
| 1ST TRIMESTER | MIDDLE TRIMESTER | 3RD TRIMESTER |
|---|---|---|
| Environment of Neural Epithelium | Laminar Hypoxia | Matrix Hemmorrhages |
| Migratory Corridors | Leukoencephalopathies | |
| Neuronal Escapes | Glial Migration | |
| Porencephaly | ||
| Multicystic Encephalomalacias | ||
| Ulegryias |
Transformation of Radial Glial Cells (RGCs)
Starting around 21 weeks of gestation, at the end of neuronal migration, RGCs undergo a transformation into astrocytes and astrocytic precursors17 (Figure 2). A radical enhancement in the abundance and activity of the lysosomal apparatus and autophagic vacuoles are observed in the RGCs, the cytological basis for resorption of the long axical process of the RGC. RGCs lose their pial attachment and their nuclei are translocated in the white matter and the lower part of the cerebral cortex. Abnormally precocious transformation of the glial guides has been observed in several circumstances included in Potter syndrome.18 Preliminary data suggest that hypoxia could modify this important part of gliogenesis.
Neuronal Displacements Outside of Migratory Corridors
Superficial ectopias, due to late neuronal escape* through holes in the limiting pialglial membrane, settle in the plexiform zone or in the leptomeningeal space. They are a constant feature of the fetal alcohol syndrome and are encountered in cases of developmental microcephaly, mental retardation of unknown origin, and localized injuries to the basal membrane or to the surface of developing brain.19 Small and isolate ectopias are occasionally encountered in otherwise normal brains. In our fetal material, these ectopias become conspicuous between 20 and 25 weeks. Marin-Padilla20 used the Golgi method to study the occurrence of neuronal ectopias in injuries to the cortical surface acquired late in gestation, associated with pial glial membrane ruptures caused by infectius, vascular, ischemic and/or hypoxic agents. He demonstrated the influence of these ectopias on the surrounding normotopic neurons.
FIGURE 2: Pattern of Distribution of Glial Cells in the Developing Neocortex: Schematic Representation
Sections in the neural tube from E8 to P8; the external and internal continuous lines represent the pial surface and the limit of the lumen (L) of the neural tube;----: limit between the germinative zone and the intermediate zone;--:limit between the intermediate zone and the cortical plate;...:limit between the infragranular cortical layers and the supragranular cortical layers. The indicated timing is valid for the murine developing brain. For clarity, the dual origin of the astrocytes is represented above (A) and below (B) the symbol of the lumen of the neural tube. These mechanisms are, of course, spatially superimposed in nature. (E8) The glial cells are regularly aligned. (E10) A and B - Before neuronal migration, the RGCs are already organized in fascicules. (E14) A - Persisting fasciculated RGC pattern during the migration of neurons destined for the infragranular layers. (E17) The stage of neuronal migration to the supragranular layers. A - Defasciculation of RGCs in the cortical plate via gradual neuronal satruation, glial dilution, and starting transformation of RGCs; B - Late multiplication and migration of glial cells from the germinative zone for the glial migration is represented within the hollow arrows. (P0) A - Gradual transformation of RGCs into astrocytes after the end of neuronal migration. (P3) A - Intense transformation of RGCs into monopolar cells in the intermediate zone. B - Appearance of mulitpolar astrocytes in the upragranular layers. (p8) Stage of complete transformation into mature glia of RGCs (intermediate zone and infragranular layers) and of late postmigratory astrocytic precursors (supragranular and marginal layers).
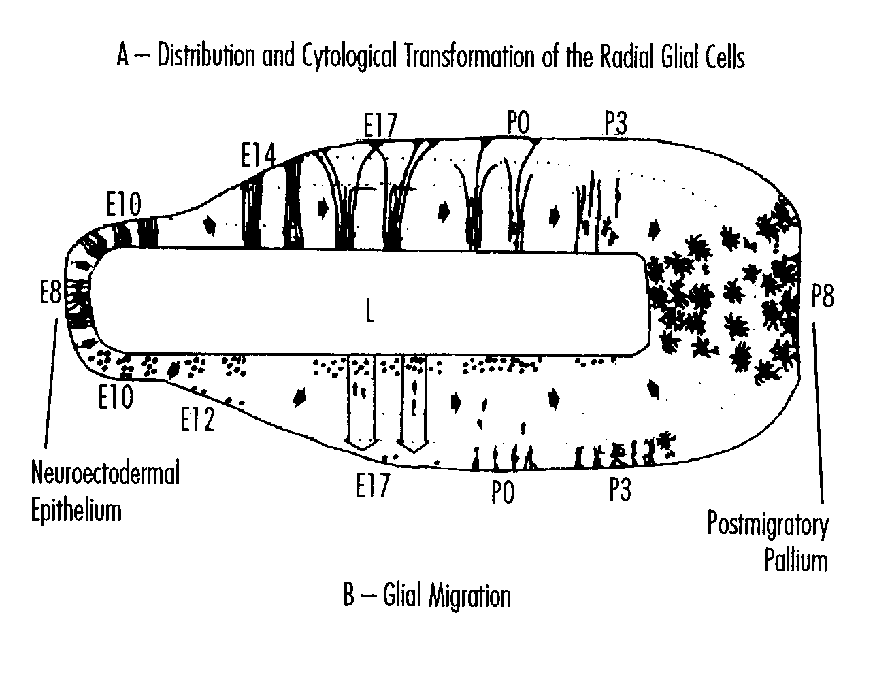
As already alluded to earlier, after neuronal destructions by ibotenic acid15 and by local freezing,21 neuronal displacements outside of the migratory corridors or along preserved RGCs are sometimes abundant enough to refill a destroyed layer with a full complement of neurons. In these neuronal compensations, the role of an adaptation of programmed cell death has not been demonstrated.
The Dual Origin of Astrocyte Precursors
Most neural cyto- and histogenetic events in the human forebrain occur during the first half of gestation. Most developmental steps in the second half of gestation launch growth and differentiation events that persist long into postnatal life. Residual, but important, histogenetic activities continue during the last 20 weeks of pregnancy. A few of them will be reviewed in some detail in this section, because of their potential importance to understanding the neuropsychological consequences of perfusion failures/hypoxia occurring during the last trimester of gestation and in premature infants.22,23
Most neurons destined for the human neocortex are produced before 20 weeks of gestation. The Perinatal Project of the National Institutes of Health provided serially processed material (Jammes and Gilles24) demonstrating that the germinative zone reaches its maximum volume around the 26th week of intrauterine life. According to these data, the conspicuous germinative zone in premature infants is either an ontogenic residue producing neurons destined to die soon after their production in or near the germinative zone, or a germinative zone destined to produce populations of glial cells. Volpe has suggested that the oligodendroglial progenitor cell, the precursor of the oligodendrocyte which forms the central nervous system myelin, is produced by the late germinative zone after the end of neuronal migration. We recently reported23 observations in the mouse suggesting that the late germinative zone produces most of the astrocytic precursors destined for the upper half of the neocortex (Figure 3). Our previous studies on gliogenesis in human fetuses17 provide indirect evidence for a similar chronology and fate of the late germinative zone in the human.
FIGURE 3: Schematic Representation of the Hypothetical Consequences of Subcortical Damage in the Premature Brain
A - Glial migration for the astrocytic equipment of the upper cortex. B- Production of the oligodendrocytes. C and D - Screens constituted by leucoencephalopathies impeding the late migration of astrological and aligodendroglial precursors. E - Hemorrhage with local destruction of the germinative zone. GZ - germinative zone; WM: white matter; CP - cortical plate. From Evrard, Gressens, and Volpe22.
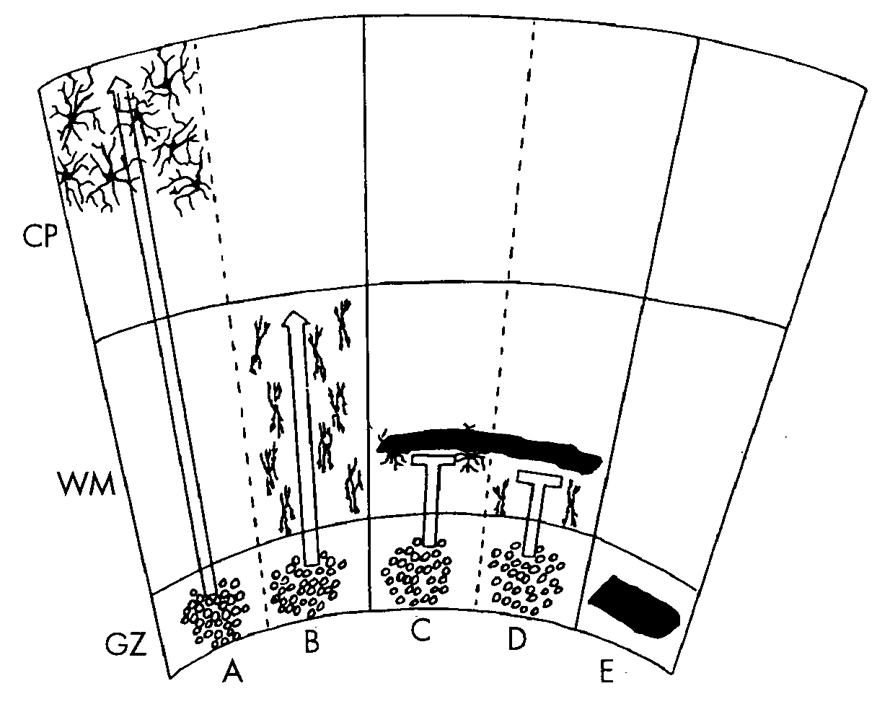
These data have implications concerning the mechanisms of brain disturbance associated with germinal matrixintraventricular hemorrhage and with periventricular leukomalacia in the premature newborn. Germinal matrix hemorrhage destroys glial precursor cells in the germinative zone. The consequences are twofold. First, oligodendroglial progenitors are eliminated and/or their migration disrupted, which may potentially result in later impaired myelination. Second, astrocytic precursors ultimately destined for the upper layers of neocortex may be destroyed and/or their migration disrupted, which may, thereby, impair cortical neuronal development, as described below. Major hemorrhagic infarction in periventricular white matter, as occurs with large, asymetric intraventricular hemorrhage, and periventricular leukomalacia clearly have major deleterious impact by interrupting projection and associative fibers. Additionally, oligodendroglia in situ, in preparation for myelination, may be destroyed with subsequent impairment of myelination. Finally, these major hemorrhagic and nonhemorrhagic lesions in the white matter may involve the neurons of the subplate which play important roles in cortical neuronal organization and in the development of cortical associative and projection connections. The importance of these mechanisms of late gliogenesis is highlighted when we consider that an appropriate astroglial contigent is a condition for the survival of a part of the neurons of the upper half of the cortex. We reported dramatic neuronal depletion (about 40 percent) in the upper half of the mouse neocortex when, after completing normal neuronogenesis, the mitotic activity of the germinative zone is inhibited during the late developmental period.23 These data also suggest a prolonged importance of Rakic's protomap for the germinative zone. These suggest that the neuronal protomap is followed by an astroglial protomap which may have a crucial role for the cytoarchitectonic integrity of the upper half of the cortex. The hypothesis that leucoencephalopathies in premature infants can act as obstacles in the pathway of late migrating glia suggests a new type of clinico- pathological correlation: the disruption of the topological and ontological relationship constituted by the radial pathway of the late produced astroglia destined for the upper half of the neocortex (Figure 2).
References
- Evrard P, Miladi N, Bonnier C, Gressens P. Normal and abnormal brain development. In: Rapin I, Segalowitz SJ (eds). Handbook of Neuropsychology, Amsterdam, 11-44, 1992.
- Gressens P, Gofflot F, Van Maele-Fabry G et al. J Neuropath Exp Neurol 51:206-219, 1992.
- Gadisseux JF, Evrard P. Glial-neuronal relationship in the developing central nervous system. A histochemical-electron microscope study of radial glial cell particulate glycogen in normal and reeler mice and the human fetus. Dev Neurosci 7:12-32, 1985.
- Gadisseux JF, Kadhim HJ, van den Bosch de Aguilar P et al. Neuron migration within the radial glial fiber system of the developing murine cerebrum: An electron microscopicautoradiographic analysis. Dev Brain Res 52:39-56, 1990.
- Gressens P, Kosofsky BE, Evrard P. Cocaine-induced disturbances of corticogenesis in the developing murine brain. Neurosci Lett 140:113-116, 1992.
- Fantel AG, Person RE, Burroughs-Gleim CJ, Mackler B. Direct embryotoxicity of cocaine in rats: Effects on mitochondrial activity cardiac function, and growth and development in vitro. Teratology 42:35-43, 1990.
- Gressens P, Lammens M, Picard JJ, Evrard P. Ethanol-induced disturbances of gliogenesis and neuronogenesis in the developing murine brain. Alcohol 27:219-226, 1992.
- Gressens P, Hill JM, Gozes I et al. Growth factor function of vasoactive intestinal peptide in whole cultured mouse embryos. Nature 362:155- 158, 1993.
- Gressens P, Cilio MR, Schlögel X, Evrard P. Les mécanismes de la souffrance cérébrale du fétus et du prématuré. Progrès en Néonatologie 11:203-226, 1991.
- Kuban KC, Gilles FH. Human telencephalic angiogenesis. Ann Neurol 17:539-548, 1958.
- Norman M, O'Kusky JR. The growth and development of microvasculature in human cerebral cortex. J Neuropathol Exp Neurol 45:222-232, 1986.
- Richman DP, Stewart RM, Caviness VS. Cerebral microgyria in a 27 weeks fetus: An architectonic and topographic analysis. J Neuropathol Exp Neurol 33:374-384, 1974.
- Barkovich AJ, Gressens P, Evrard P. Formation, maturation, and disorders of brain neocortex. Am J Neuroradiol 13:423-446, 1992.
- Evrard P, Kadhim HJ, de Saint-Georges P, Gadisseux JF. Abnormal development and destructive processes of the human brain during the second half of gestation. In: Evrard P, Minkowski A (eds). Developmental Neurobiology. New York: Raven Press, 21-41, 1989.
- Innocenti GM, Berbel P. Analysis of an experimental cortical network: I) Architectonics of visual areas 17 and 18 after neonatal injections of ibotenic acid; similarities with human microgyrias. J Neural Transplant Plast 2:1-28, 1991.
- Caviness VS, Misson JR, Gadisseux JF. In: Galaburda A (ed). From Reading to Neurons. Cambridge: M.I.T. Press, 404-442, 1989.
- Kadhim HJ, Gadisseux JF, Evrard P. Topographical and cytological evolution of the glial phase during the prenatal development of the human brain: A histochemical and electron microscopic study J Neuropathol Exp Neurol 47:166-188, 1988.
- Kadhim HJ, Lammens M, Gosseye S et al. Brain defects in infants with Potter syndrome (oligohydramnios sequence). Pediatr Pathol 13:519-537, 1993.
- Caviness VS, Evrard P, Lyon G. Radial neuronal assemblies, ectopia and necrosis of developing brain. Acta Neuropathol 41:67-72, 1978.
- Marin-Padilla M. Leptomeningeal heterotopias and cortical architecture reorganization: A Golgi study. In: Galaburda A (ed). The Extraordinary Brain. (In press.)
- Dvorak K, Feit J. Migration of neuroblasts through partial necrosis of the cerebral cortex in newborn rats—contribution to the problems of morphological development and developmental period of cerebral microgyria. Acta Neuropathol 38:203-212, 1977.
- Evrard P, Gressens P, Volpe JJ. New concepts to understand the neurological consequences of subcortical lesions in the premature brain. Biol Neonate 61:1-3, 1992.
- Gressens P, Richelme C, Kadhim HJ et al. The germinative zone produces most cortical astrocytes after neuronal migration in developing mammalian brain. Biol Neonate 61:4-24, 1992.
- Jammes JL, Gilles FH. Telencephalic development: matrix volume and isocortex and allocortex surface area. In: Gilles FH, Leviton A, Dooling EC (eds). The Developing Human Brain—Growth and Epidemiologic Neuropathology. Boston: J Wright PSG Inc., 87-93, 1983.
Discussion
Ingemar Kjellmer, M.D., Ph.D.
Gothenburg University, Goteburg, Sweden
With the wealth of information that has been given in the previous talks I will have to restrict myself to comment on only a few of the subjects presented. I shall use my time first to ask Dr. Gluckman about his time schedule for brain injury, second to present some new data to reinforce the claim that oxygen derived free radicals are at play and that there is evidence in humans that perinatal asphyxia may lead to neuronal destruction. Third, I want to endorse the idea that excitatory amino acids may be released also from human brains after perinatal asphyxia. Finally, as a short comment to Drs. Parer and Greisen I will discuss oxygen extraction of the brain when oxygen lack and acidosis coincide.
First then, the time schedule: I find it quite useful and necessary to make a clear distinction between primary neuronal death and secondary neuronal death. But I would like to ask Dr. Gluckman to elaborate on the advantage of subdividing the neuronal loss that takes part after reoxygenation or reperfusion has started (from the neonatologist point of view—after start of resuscitation) into reactive cell death or reperfusion injury and delayed neuronal death. To my understanding it is not entirely clear that reperfusion injury is a matter of shortlasting events but rather that these two types of processes may well extend over unknown periods of time.
After experimentally induced ischemia Bromont et al.1 demonstrated prolonged augmentation of lipid peroxidation over a period of more than 72 hours and the release of excitatory amino acids that Dr. Johnston has described may also have effects lasting over a certain period of time, for instance via the activation of the phosphoinositol second messenger system.
The time schedule naturally is necessary for the discussion of a therapeutic window.
Then over to the free radicals alluded to by both Dr. Gluckman and Dr. Johnston. Because of their extreme reactivity and short biological half lives they are characteristically elusive by nature. We have had to be content to study the traces that they leave and the effect of scavenging. However, by means of spin trapping, the unpaired electron of the free radical may be trapped by a material which itself becomes a radical. When a substance is chosen that forms stable radicals, the possibility of quantifying the amount of radicals produced by the use of electron magnetic resonance spectrometry is at hand. I will very briefly describe to you a study which we recently completed.3 The spin trap methodology was applied using OXANOH as the radical forming adduct OXONO· to study the formation of free radicals in the brain of the anesthetized fetal sheep during ischemia/reperfusion. We took advantage of the preparation described by Dr. Gluckman and coworkers2 to ligate the communications between the vertebral and carotid arteries, thus creating a situation where the brain is supplied solely from the carotids. OXANOH—in its reduced, non-radical form—was infused intra-arterially to the brain via one lingual artery. Blood samples were collected from sagittal sinus blood and analyzed for OXANO· —the stable free radical form—using electron spin resonance spectroseopy After a control period, a 30-minute period of bilateral occlusion of the carotid rendered the brain ischemic. During recirculation a significant increase in the amount of radicals liberated from the brain was demonstrated, peaking at 10 minutes of reperfusion and lasting for at least one hour (Figure 1). This is to my knowledge the first direct demonstration of the increased generation of free radicals in the perinatal brain during reperfusion.
FIGURE 1: Production of Free Radicals in Fetal Brain
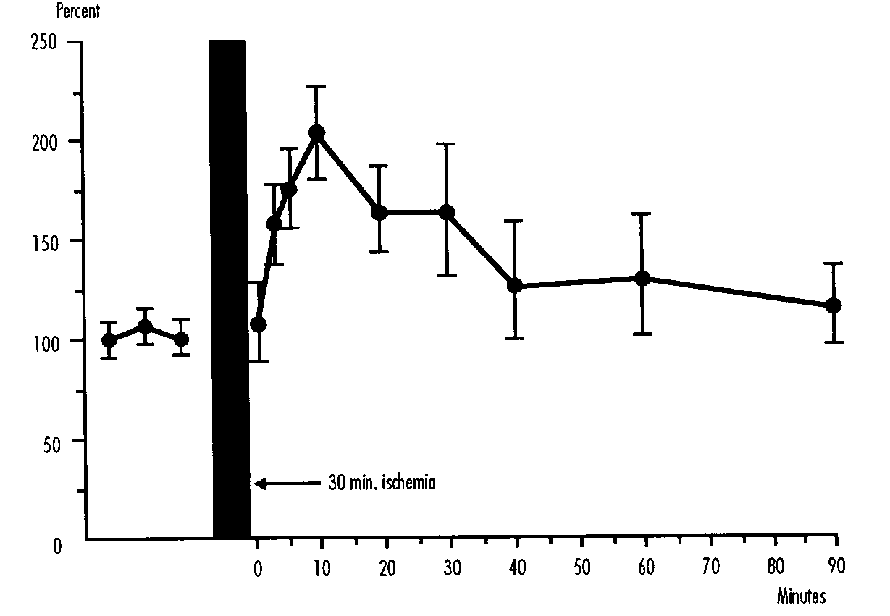
FIGURE 2: Neuron Specific Enolase in CSF and Serum after Asphyxia
Concentrations of neuron specific enolase (NSE) in serum and CSF in four groups of neonates: healthy control babies, and neonates resuscitated for birth asphyxia and developing mild, moderate and severe hypoxic-ischemic encephalopathy.
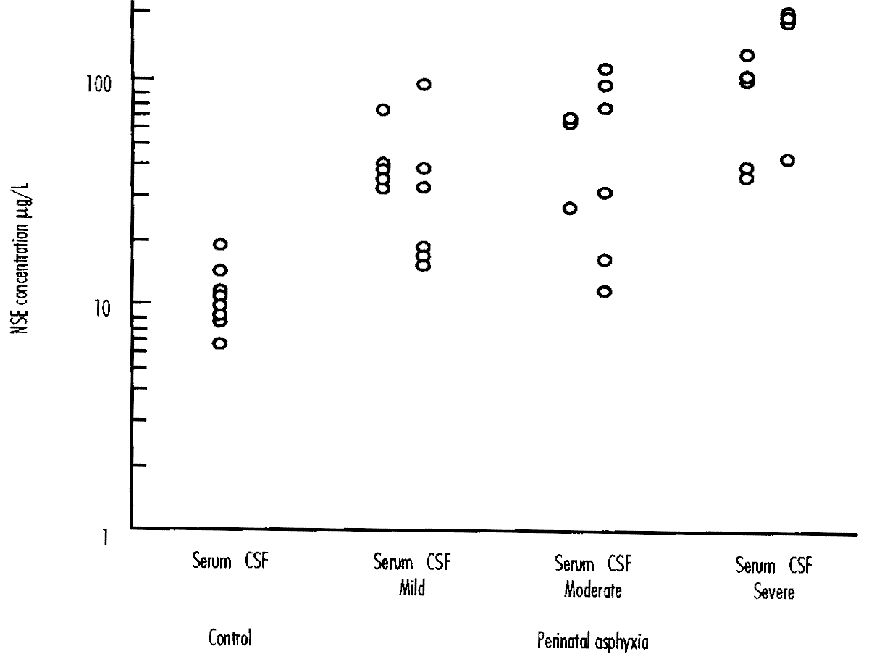
Neuron Specific Enolase in CSF and Serum After Asphyxia
What evidence do we have that perinatal asphyxia in humans actually causes neuronal destruction? We collected CSF samples from 16 newborn infants during their first day of life.4 Six had a mild, six a moderate and four a severe hypoxic-ischemic encephalopathy (HIE). Nerve specific enolase (NSE) was measured in CSF and serum (Figure 2). All three groups of asphyxiated babies had higher NSE levels than eight control babies. In neonates with clinical signs of severe HIE NSE values in CSF were very high and individually higher in CSF than in serum, demonstrating a leakage of the enzyme from CNS.
I then want to turn to the excitatory amino acids. In a similar series of asphyxiated human infants we studied the concentration of amino acid in the CSF (Figure 3). Glutamate and aspartate concentrations were elevated, especially so in babies who developed clinical signs of severe encephalopathy.5
Finally, I want to comment on the presentations of Dr. Parer and Dr. Greisen on the blood flow and oxygen consumption of the fetal brain. It is apparent that in both humans and sheep oxygen consumption of the brain is very low during midgestation, increases at full term but still at this stage does not reach the same levels as in the adult. A drastic illustration of extremely low values for oxygen consumption in human neonatal brain was recently given by Altman et al.6 They suggest that at least occasional extremely low levels for oxygen uptake may be compatible with intact survival. Dr. Parer stresses the point that oxygen delivery to the fetal brain exceeds that of the adult suggesting a reserve blood flow. I would propose an alternate mechanism. In studies on the exteriorized fetal lamb that were performed 20 years ago7 we demonstrated a peculiarity of the relation between oxygen delivery, oxygen consumption and acidosis (Figure 4). When fetal lambs were rendered hypoxic and simultaneously pH was manipulated—either by infusion of bicarbonate to the fetus or by CO 2 breathing by the maternal ewe—it became apparent that the brains of the fetuses that were acidotic (filled or half-filled symbols) had a considerably lower oxygen consumption than fetuses with no or only mild acidosis. In other words, despite the fact that CBF was the same (105 ml/min/100g) and the arterial saturation was the same, i.e., oxygen delivery to the brain was the same in the acidotic and the non-acidotic group, oxygen extraction was significantly reduced in the acidotic fetuses. So I suggest that when high cerebral blood flow is observed in the fetus or neonate it might signal poor oxygen extraction in connection with acidosis.
FIGURE 3: Concentration of Amino Acid in CSF of Asphyxiated Human Newborns
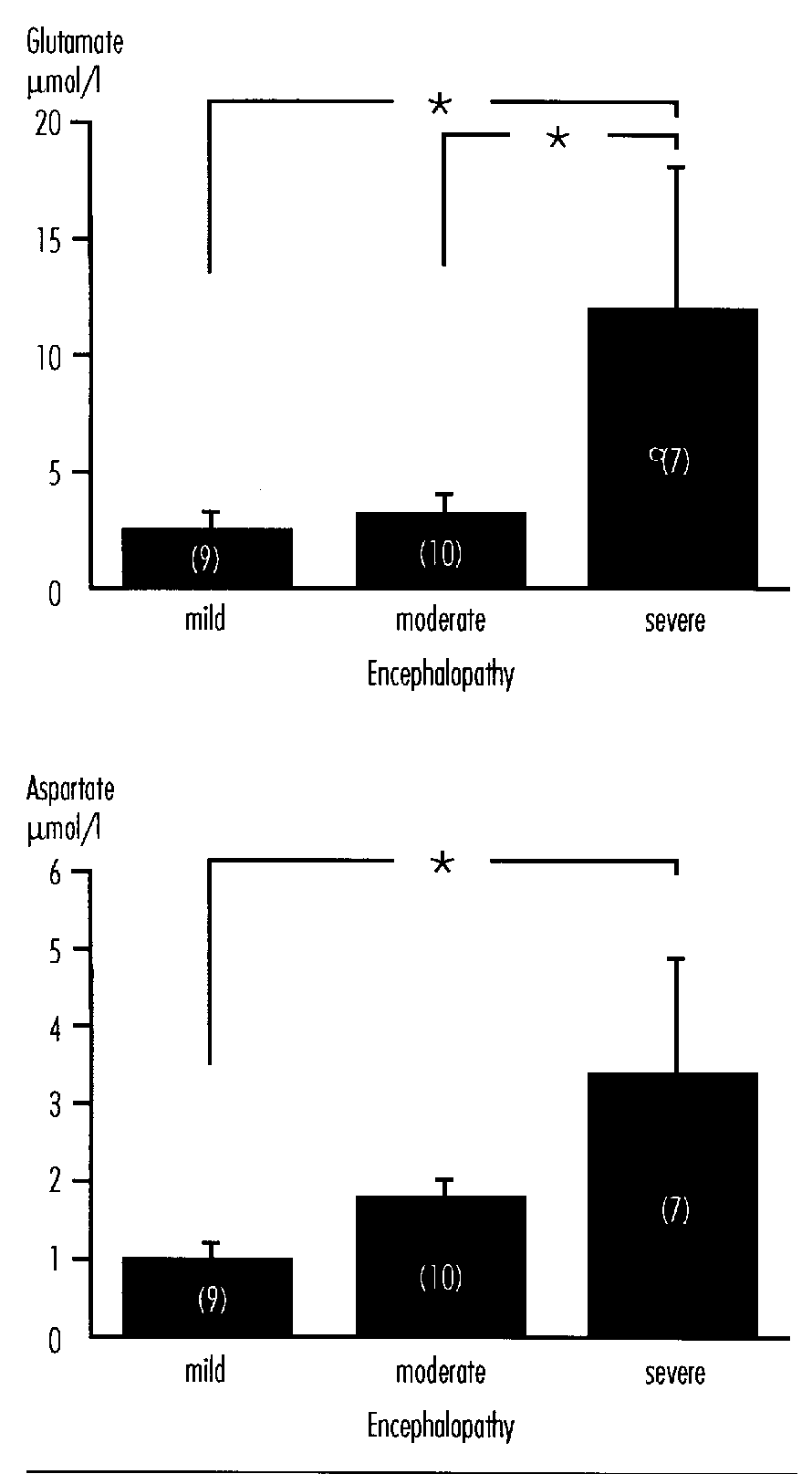
FIGURE 4: Relation Between Oxygen Delivery, Oxygen Consumption and Acidosis in Fetal Lambs
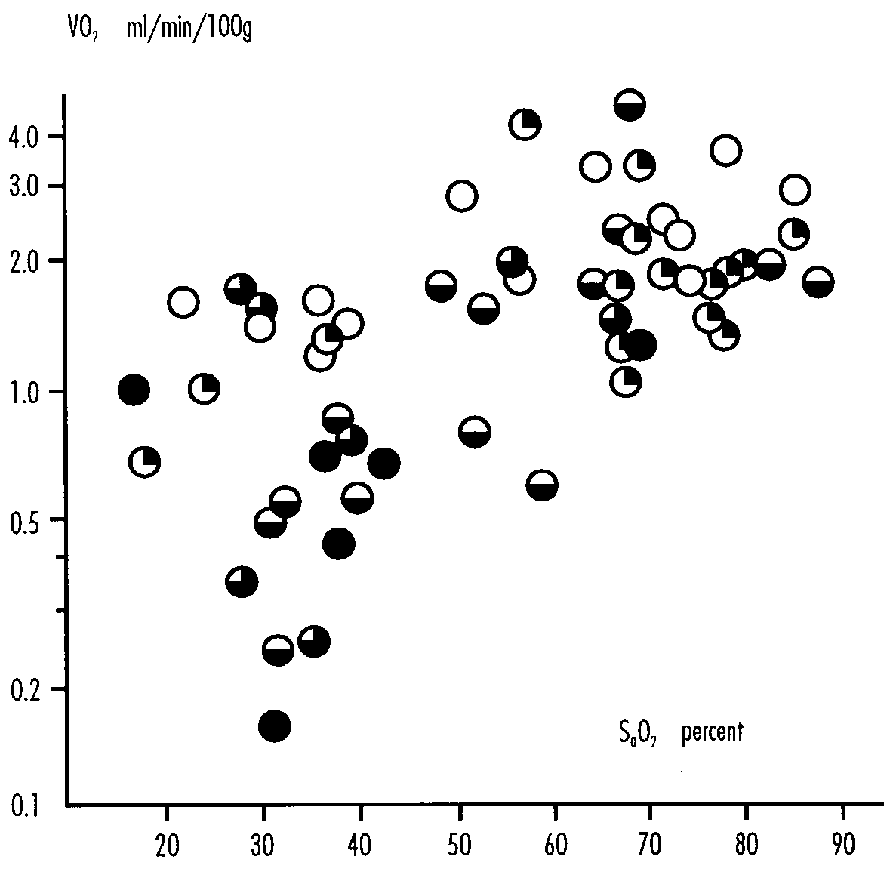
References
- Bromont C, Marie C, Bralet J. Increased lipid peroxidation in vulnerable brain regions after transient forebrain ischemia in rats. Stroke 20:918-924, 1989.
- Williams CE, Gunn AJ, Gluckman PD, Synek B. Delayed seizures occurring with hypoxic-ischemic encephalopathy in the fetal sheep. Pediatr Res 27:561-565, 1990.
- Bägenholm R, Nilsson UA, Wallin Götborg C, Kjellmer I. Free radicals are formed during reperfusion in the brain of fetal sheep. 14th European Congress of Perinatal Medicine, Helsinki, Kirjapaino Tapo Oy, 1994: A283.
- Thornberg E, Thiringer K, Hagberg H, Kjellmer I. Neuron specific enolase in asphyxiated newborns. Arch Dis Child 72:F39-F42, 1995.
- Hagberg H, Thornberg E, Blennow M et al. Excitatory amino acids in the cerebrospinal fluid of asphyxiated infants: Relationship to hypoxic- ischemic encephalopathy. Acta Paediat 82:925-929, 1993.
- Altman DI, Perlman JM, Volpe JJ, Powers WJ. Cerebral oxygen metabolism in newborns. Pediatrics 92:99-104, 1993.
- Kjellmer I, Karlsson K, Olsson T, Rosen KG. Cerebral reactions during intrauterine asphyxia in the sheep. I. Circulation and oxygen consumption in the fetal brain. Pediatr Res 8:50-57, 1974.
Discussion
Philippe Evrard, M.D.
Service de Neurologie Pédiatrique, Hôpital
Universitaire Saint-Luc and Laboratoire de
Neurologie du Développement, University
of Louvain Medical School at Brussels, Belgium
The speakers in this session, who are renowned leaders in the field, provided superb contributions reviewing their own original data as well as the literature. In order to stimulate the discussion and to contribute to the consensus definition and to the research planning which are the main purposes of this NIH meeting, I shall summarize and discuss as a whole the material submitted for the first session of this meeting without giving a detailed separate credit to each contributor. I shall make no effort to be exhaustive in the discussion of such vast and rich material. Because it is more provocative for the discussion, I shall mix in my comments the data and concepts reported in the manuscripts with those from other sources.1-11
I remind the group that one of the purposes of this first session is basic preparation to answer the following questions:
- Does a consensus definition now exist?
- If not, what research is needed to develop one?
- Is research needed to validate a research definition?
- Is research needed to assess relation to outcome:
- short term?
- long term?
Summary of the Papers and Comments
From the four submitted documents, from the whole literature, and from the selection of the speakers by the organizers of this meeting, it appears, and I very much appreciate, that there is a desirable continuum in the study of all aspects of brain injury from the time of conception until the end of the neonatal period. Birth is a specific event, which carries for the fetus transforming into a newborn the following dangers and many others: changes in the arterial blood pressure, establishment of the respiratory function, stress, humoral modifications, inappropriate treatment and inappropriate care, transitory loss of cerebral auto-regulation. It could be a reason for separating brain injury at birth from brain injury at any other developmental step. But there are several reasons to support the continuity in studying brain injury throughout the developmental span.
- Dr. Karin Nelson's epidemiological data establish that prenatal and preconceptional factors play a major role in cerebral disabilities. Prenatal brain hypoxia-ischemia and prenatal events favoring neonatal hypoxia-ischemia are among the prenatal factors in the etiopathogenesis of cerebral disabilities which will be reviewed by Dr. Karin Nelson during this meeting.
- Several socioeconomical problems which favor the occurrence of brain hypoxia are the same during pregnancy, at birth and during the neonatal period.
- The transfer of information about the patients from obstetricians to neonatologists and pediatric neurologists is most important for the patients and their parents. The continuum of care from fetal to postnatal medicine is a crucial priority.
- Several cellular alterations associated with brain asphyxia are similar during the second half of gestation, at birth, and during the first months of postnatal life. Similar treatments and prophylactic measures against these neuronal and glial alterations are under consideration for the pre- and postnatal periods.
- The hypoxia opportunism, as I called it, exploits the developmental mechanisms at most consecutive developmental steps.1,2 Research strategies will benefit from a comprehensive approach to these steps.
The amount of chemical compounds available to the neurons and glial cells (oxygen, glucose, other nutriments) depends upon the cerebral blood flow, upon the concentrations of these compounds in blood, and upon the multiple barrier systems between blood and neural elements3. Hypoxias/ischemias/perfusion failures can provoke brain damage when the transfer of compounds from blood to neural tissue is reduced to a level under the possible compensations. The clearance of compounds (CO 2 and others) from neural tissue to blood is also impaired by ischemias/perfusion failures. Dr. Greisen and Dr. Parer focused on these questions. A multitude of data concerning these transfers and their parameters have been reported and discussed in the literature during the last two decades. In addition to their own original data, the authors of the reports provide critical and enlightening analyses which draw clear conclusions from this most complex literature.
- Oxygen consumption by fetal sheep brain is remarkably constant in conditions of hypoxia: cerebral blood flow (CBF) increases to match the reduction in oxygen blood content. Regulation of hypoxia-associated vasodilation involves several factors, among them: O2, CO 2, adenosine, prostaglandins, arginine, vasopressine. When asphyxia becomes severe, CBF and oxygen consumption fall. Oxygen uptake in fetal sheep can be reduced by 50 percent before the occurrence of evident neuronal damage. When oxygen is limited, fetal brain can readily use anaerobic glycolysis which produces lactate. Lactate efflux from the brain is slow and high lactate levels in brain can be implicated in neuronal damage.
- On the basis of a few exceptional observations in the human newborn and on the basis of extrapolations from other mammalian newborns, it is assumed that CBF in acute hypoxemia in human newborns may increase twofold to threefold by vasodilation, provided perfusion pressure is maintained. This allows electrical function (electrical failure is a prewarning of permanent tissue injury) to persist until arterial oxygen saturation falls below 50 percent. However, increased oxygen extraction may decrease venous oxygen saturation from normal levels of 65-70 percent to a critical level (40-45 percent as an average, but this percentage can be modified by alkalosis, high fraction of fetal hemoglobin) which corresponds to the minimal oxygen tension providing a sufficient diffusion gradient from the venous end of the capillary to the mitochondria of the most deprivileged neuron. In asphyxia of non-human mammalian newborns (or perinatal animals), a redistribution within the brain (luxury perfusion) has been constantly demonstrated, mediated through the sympathoadrenergic system. In human newborns, a state of delayed luxury perfusion with lost reactivity has been documented, which carries a poor prognosis. It is, however, unclear if this cerebrovascular disturbance (delay of the luxury perfusion) is a cause or a consequence of neuronal injury.
- The heterogeneity of hypoxic-ischemic thresholds becomes an important issue when discussing CBF, oxygen, and substrates in brain injuries. I shall quote a summary of Dr. Vannucci's contributions about this question.4-6 "Hypoxemia of a severity potentially adequate to produce brain damage is always associated with substantial cardiac depression leading to systemic hypotension and secondary cerebral ischemia; acidosis is a major contributing factor. Thus, cerebral ischemia occurs as a consequence of hypoxic-acidotic cardiovascular depression or primarily as in occlusive vascular disease. lschemia leads not only to a tissue oxygen debt, but also to a substrate (glucose) debt; the combination is especially harmful to the immature brain with its limited capacity for glucose transport from blood into neurons and glia. The immature brain is more resistant to the deleterious effect of hypoxia-ischemia. Perinatal animals of several species (sheep, dogs) require at least 15 minutes of total anoxemia or asphyxia for brain damage to occur, and some species (rats, rabbits) tolerate 25 or more minutes. Monkeys tolerate 8-12 minutes. At the other end of the spectrum is partial hypoxemia or asphyxia in which brain damage is not initiated during the first 1-3 hours or never occurs, owing to the fact that the systemic hypoxia-acidosis is not severe enough to produce the systemic hypotension and secondary cerebral ischemia necessary to produce tissue necrosis. In this regard, the threshold of arterial blood pressure below which damage occursapproximates one-third of the normal blood pressure. The lower the pressure is below the threshold value during the hypoxemia, the shorter is the interval before brain damage appears."
Dr. Gluckman and Dr. Johnston reviewed the cellular alterations associated with brain asphyxia, especially during the perinatal period. Asphyxia triggers a cascade of cellular biochemical events that lead to temporary alterations in cellular function and/or cell death. The excellent schematic representations proposed by Johnston and by McDonald and Johnston7 outline the numerous neurochemical and pharmacological tracks and pathways which have been demonstrated or hypothesized. The following concepts, made on the basis of the circulated material from Gluckman and from Johnston, should be emphasized:
- Distinctions between primary loss, reactive cell death or reperfusion injury, delayed neuronal death;
- Sensitizing factors: gestational age, IUGR, metabolic factors, brain temperature, pattern of injury;
- Distinction between selective neuronal death/apoptosis and infarction; and
- Mechanisms exceeding the basic cascade of biochemical events due to ischemia: loss of endothelial integrity, role of seizures, macrophage and microglial activation, several aspects of apoptosis.
Gluckman and Johnston insist on different aspects, possible implications and strategies for prevention and management:
- Current Management. Blood pressure, glucose level, vigorous anti-seizure therapy, prevention of massive edema.
- Endogenous Protective Mechanisms. Inhibitory neuromodulators, hypothermia, immune modulators, neurotrophic factors, microvascular factors. Among the possible future therapies (and, for several drugs, first results): glutamate antagonists, calcium antagonists, drugs that alter intracellular calcium concentrations and others that improve cellular energy metabolism.
- Key Factors for Planning of Clinical Trials. Definition of time of intervention, role of sensitizing factors, nature of the injury.
- Strategies.
- Primary neuronal death— prophylactic therapies
- Reperfusion phase—action before or during the insult
- Delayed cell death—neurotrophic agents against apoptosis, salvaging agents as excitatory amino acids antagonists, immunomodulators, post- asphyxial seizures
Reading the papers given by the speakers in this session with my eyes of developmentalist (see the enclosed review and discussion about Hypoxia Opportunism During Brain Development), I wish to suggest research about the modulated application of the neurochemical data and of hemodynamic pathogenesis (including vascular physiology, cerebral blood flow, etc.) to all the different finesteps of the developmental program and events. For instance, cerebral blood flow studies are most needed, but we need to take into account the precise time features and regional aspects of angiogenesis and of the metabolism of the different cell types in order to apply and to benefit fully from the data about blood flow. I wish also to underline how important it is to try to apply the general data to the different populations of cells (neurons, glia, different types of elements in the glial phase), and to use the multiple and sophisticated blood brain barriers described by Mollgard.3 A minority component (i.e., the radial glial cells) can have, at a given time, a major morphogenetic role which is not clear unless we apply the general metabolic data in a specific way. New brain imaging methods can help us very much for this difficult application of general parameters at the microscopic level.
Basic aspects of brain injury are not so distant from epidemiological data. The concept and the observations made by Dr. Karin Nelson that many, and probably the majority of cerebral palsy (CP) and cerebral disabilities, are from prenatal and/or preconceptional origin are among the great contributions of our decade and Dr. Nelson's views will surely have (and have already) an important beneficial role on medical practice of obstetricians, neonatologists, pediatricians and pediatric neurologists. Karin Nelson's epidemiological observations also facilitated our neurobiological and neuropathological research. In our own material of fetal neuropathology, we often find solid evidence supporting the same conclusions and, providing insight into many underestimated or neglected signs of fetal disorders able to provoke neonatal problems and long term neurological handicaps, directly or through secondary neonatal problems. In the discussion of this concept and of its bases, I wish to add several points:
- When we read Dr. Nelson's contributions about preconceptional and prenatal etiological factors of CP, as clinicians, neuropathologists, and neurodevelopmentalists, we are delighted to obtain through Dr. Nelson's discoveries a vast field of possible pathogenetic mechanisms able to express themselves at many developmental stages from conception until the postnatal life (and the late synaptic stabilization epoch). The original etiology can be early with comparatively late and multi-factorial pathogenetic applications: it is a consequence of the complexity and long-term planning of the neurodevelopmental program. I have the impression that several colleagues oversimplify the application of the epidemiological contribution in the following sentence: "every lesion before birth." It could lower the insurance fees but it would not be good science. A PVL occuring after birth could still be the long term consequence of a prenatal and even preconceptional etiological factor and in certain cases could still be prevented from occurring with appropriate therapeutic strategies applied by neonatologists.
- During the last few years, we read in the introduction section of many papers and of manuscripts sent to referees that "all PVL and almost all brain injuries discovered in the neonatal period are from prenatal origin." Superb contributions, for instance by Weindling8 demonstrates very elegantly that PVL can be of prenatal origin.* Jonathan Wigglesworth,9 several other neuropathologists and myself, discussed recently and compared our PVL and other perinatal brain injury material. We think that, besides their full occurrence and, in other cases, their preparation in the prenatal period, PVL continues to occur often in the neonatal, postnatal, perinatal periods.
- We recommend that neuropathological efforts continue to be supported in order to assess in the future the results of new treatments and therapeutic strategies. At an epoch when trained neuropathologists become rare and get adequate support only for biopsies and surgical procedures, it would be dangerous to neglect, without adequate proof, the neonatal and perinatal events. Among the research priorities in developmental neuropathology:
- PVL is a crucial issue in brain asphyxia in obstetrics and neonatology. The idea that PVL "occurs in a vulnerable area between the white matter supplied on the one hand by the cortical vessels and on the other by branches of vessels running in the ependyma" derives from excellent papers, including the classical papers by Vander Eecken and Adams, by DeReuck and Richardson. This view is now somewhat controversial, especially after the papers by Norman and O'Kusky and by Kuban and Gilles. The PVL problem has to be revisited.
- The hypothesis that ischemic-hypoxic leucoencephalopathies in premature infants can interfere with an astroglial protomap for the upper cortex15 deserves to be confirmed: it could open a new type of clinicopathological correlation in brain asphyxias.
- A special initiative should be undertaken with the new neuropathological methods which provide information on timing and pathogenesis, and in modern neuropathology, with its powerful joint ventures with genetics, chemical and regional neuroanatomy.
- Development of sophisticated fetal neuropathology (with the appropriate ethical precautions) seems a priority.
- Comparisons between the different medical, social, and health care delivery systems existing in our developed countries become a practical and a scientific need. For instance, most papers which appear about prognosis of prenatal and neonatal conditions and about therapeutic results are not at all applicable from U.S. to WesternEurope (and the reverse is true also).
- At a recent Little Foundation meeting,16 the occurrence of infection during the middle trimester of gestation was reported to be grossly underestimated and neglected. It could be a crucial prenatal etiological factor of CP. This point overlaps the issue of prenatal asphyxia and could deserve a better ranking in the research planning.
References
- Gressens P, Cilio R, Schlögel X, Evrard P. Les mécanismes de la souffrance cérébrale du fétus et du prématuré. Progrès en Néonatologie 11:203-226, 1992.
- Evrard P. Environmental determinants of prenatal nervous development, with particular emphasis on hypoxic-ischemic pathology. In: Brain Lesions in the Newborn: Hypoxic and Haemodynamic Pathogenesis. Lou HC, Greisen G, Larsen JF (eds). Alfred Benzon Symposium 37. Copenhagen: Munksgaard, 1994.
- Mollgard K. Barrier systems and growth factors in the developing brain. In: Brain Lesions in the Newborn: Hypoxic and Haemodynamic Pathogenesis. Lou HC, Greisen G, Larsen JF (eds). Alfred Benzon Symposium 37. Copenhagen: Munksgaard, 1994.
- Vannucci RC. Experimental biology of cerebral hypoxia-ischemia: Relation to perinatal brain damage. Ped Res 27:317-326, 1990.
- Vannucci RC. Heterogeneity of hypoxic-ischemic thresholds in experimental animals. In: Brain; Lesions in the Newborn:Hypoxic and Haemodynamic Pathogenesis. Lou HC, Greisen G,Larsen JF (eds). Alfred Benzon Symposium 37. Copenhagen: Munksgaard, 1994.
- Vannucci RC, Christensen MA, Stein DT. Regional cerebral glucose utilization in the immature rat: Effect of hypoxia-ischemia. Ped Res 26:208 -214, 1989.
- McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41-70, 1990.
- Weindling AM. Prenatal pathologic events vs periventricular leucomalacia and periventricular hemorrhage. In:Brain Lesions in the Newborn: Hypoxic and Haemodynamic Pathogenesis. Lou HC, Greisen G, Larsen JF (eds). Alfred Benzon Symposium 37. Copenhagen: Munksgaard, 1994.
- Wigglesworth JS. Neuropathological clues to the timing of early brain lesions. In: Brain Lesions in the Newborn: Hypoxic and Haemodynamic Pathogenesis. Lou HC, Greisen G, Larsen JF (eds). Alfred Benzon Symposium 37. Copenhagen: Munksgaard, 1994.
- Evrard P, Gressens P Volpe JJ. New concepts to understand the neurological consequences of subcortical lesions in the premature brain. Biol Neonate 61:1-3, 1992.
- Bax M. The middle trimester of gestation and etiologies of cerebral palsy. Little Foundation meeting. Oxford, U.K.: Castle Priory, January 1993.
Session II: Clinical Assessment— Obstetrics
Moderator: Edward J. Quilligan
Fetal Monitoring: Utility and Interpretation of Umbilical Cord Blood Gases and Fetal Scalp Sampling
John C. Hauth, M.D.
Department of Obstetrics and Gynecology
Division of Maternal–Fetal Medicine
University of Alabama at Birmingham
Background
In 1861 Little described a correlation between pregnancy, labor and delivery complications and subsequent long term neurologic deficits.1 Thereafter for more than a century, it was commonly believed that most developmental problems in children were attributable to events surrounding delivery. Specifically, peripartum asphyxia was commonly believed to be the primary cause of cerebral palsy, mental retardation, and other forms of neurological dysfunction. The misperception that birth asphyxia accounts for a significant portion of infants with cerebral palsy continues to exist, despite the infrequent association of intrapartum events and subsequent cerebral palsy. However, a 1985 National Institutes of Health report2 concluded that the association of intrapartum events and subsequent adverse neurologic effects (including cerebral palsy) is infrequent. More recently, a number of investigators have confirmed that few cases of cerebral palsy and other developmental impairments can be associated with events which occur during the delivery process.3-6 For example, Nelson and Ellenberg4 using data from the Collaborative Perinatal project, reported that events occurring during labor and delivery accounted for only a small proportion of the total cases of cerebral palsy.
Thus, these reports confirm that the cause(s) of cerebral palsy are not known but that the overwhelming association is with extreme prematurity, maternal mental retardation and anomalous infants. In term gestations less than 10 percent of subsequent cerebral palsy has been associated with intrapartum events. Despite new obstetric and neonatal technologies, including intrapartum fetal heart rate pattem assessment7 to identify fetal hypoxia that has contributed to the marked increase in cesarean deliveries in the United States, in Western industrialized counties, the rate of cerebral palsy in term infants (1-2/1,000) has remained unchanged over 20 years.8
However, for academic and medical-legal considerations it has become important to more precisely define birth asphyxia. In the absence of newborn metabolic acidemia it is a physiologic certainty that proximate fetal hypoxia did not occur. Therefore, we must determine what severity of newborn metabolic acidemia, a definite sequelae of substantial fetal hypoxia, is associated with neonatal seizures, prolonged hypotonia or multiorgan system dysfunction. In the absence of these conditions it is reasonable to conclude that proximate intrapartum management, that did not result in severe newborn metabolic acidemia, is an unlikely cause of subsequent adverse neurologic events. The American College of Obstetricians and Gynecologists and the American Academy of Pediatrics have challenged the use of the Apgar score to define birth asphyxia.9 The Apgar scoring system, first devised in 1952, has been used to assess newborn condition and to reflect the need for resuscitation. Over time, the Apgar score has also been used to define asphyxia, which is inappropriate, as many other conditions (e.g., congenital anomalies, prematurity, maternal drug administration) can result in low scores that are not reflective of asphyxia. Asphyxia implies hypoxemia to a degree leading to metabolic acidosis and cannot establish the occurrence of sufficient fetal hypoxia to cause subsequent cerebral palsy. An infant with an Apgar score of 0-3 at 5 minutes whose 10 minute score improves to 4 or better has a 99 percent chance of not having cerebral palsy at 7 years of age. It should also be noted that 75 percent of children in whom cerebral palsy subsequently developed had normal Apgar scores at birth.10
Umbilical cord blood acid-base assessment is a more objective measure of the status of a newborn than are Apgar scores. Knowledge of fetal physiology, acid-base regulation and response to acute or chronic hypoxic stress should allow practitioners to utilize umbilical cord blood gas values to assess the appropriateness of intrapartum management. It is not accurate to simply define birth asphyxia as any degree of metabolic acidemia presumed due to fetal hypoxia. The terms fetal distress and asphyxia have been variously applied to newborns with mild umbilical artery acidemia, all labors complicated by meconium-stained amniotic fluid, newborns with a low 1- or 5-minute Apgar score, or, most commonly, with an abnormal or nonreassuring fetal heart rate pattern. These usages are nonspecific, imprecise, and have a low predictive value for immediate newborn complications or long-term adverse neurologic sequelae attributable to substantial fetal hypoxia. A fetus who has suffered hypoxia proximate to delivery that is severe enough to result in hypoxic encephalopathy will show other evidence of hypoxic damage including all of the following.11
- A profound umbilical artery metabolic or mixed acidemia (pH <7.00).
- Persistence of an Apgar score of 0-3 for longer than 5 minutes.
- Neonatal neurologic sequelae, e.g., seizures, coma, hypotonia.
- Multiorgan system dysfunction, e.g., cardiovascular, gastrointestinal, hematologic, pulmonary or renal.
The term birth asphyxia is imprecise and should not be used.
Physiologic Considerations
The fetus is dependent on placental exchange for oxygen, nutrients, and the transfer of waste products. This exchange takes place in the intervillous space where maternal blood bathes the placental villi, which are rich in fetal capillaries. Transfer in both directions occurs across this semipermeable membrane.
Labor is a stress to the fetus. Uterine contractions impede blood flow in the intervillous space to a degree proportional to the duration, intensity, and frequency of the contractions. Thus, each contraction briefly interrupts the supply of oxygen to the fetus. Moreover, compression of the umbilical cord, with or without contractions, interrupts the flow of oxygen to the fetus. The normal umbilical venous P02 averages 28-30 mmHg. This very low oxygen tension is well tolerated by the healthy fetus because of the increased fetal hemoglobin concentration and the increased fetal heart rate of 120-160 beats/min. The binding of 02, H+, and CO2 are different in the fetus and placenta compared with the adult because of the differences in acidity in fetal blood and the capabilities of fetal hemoglobin. This Bohr effect allows more efficient oxygen uptake in the placenta and more efficient oxygen release in the fetal tissues. The fetal basal metabolic rate and hence oxygen requirement is also markedly lower than that of the newborn. Normal fetuses generally tolerate brief interruptions of their oxygen supply well. Interruption of nutrient transport is tolerated for even longer periods because of the storage capabilities of the fetus and placenta.
Normally, the fetal life is maintained through oxidative metabolism. Volatile acids diffuse readily across the placenta into the maternal circulation, thus maintaining fetal homeostasis. During periods of diminished oxygen supply the fetal metabolism may convert to an anaerobic pathway (Embden-Meyerhof), with the formation of organic acids such as lactic acid. These organic acids diffuse slowly across the placenta. Nevertheless, fetal buffering systems can maintain a normal pH for a period of time. However, maternal acid-base buffers (such as bicarbonate) diffuse slowly across the placenta and CO2 diffuses rapidly. Thus, prolonged interruption of the fetal oxygen supply will eventually result in metabolic acidosis in the fetus. Chemoreceptors and baroreceptors in the fetus are responsive to changes in pH, PCO 2, and P02. These receptors regulate the fetal circulation and can alter fetal heart rate patterns. Unfortunately, these altered fetal heart rate changes are frequently nonspecific and have a poor correlation with both immediate and long-term neurologic outcomes.
A healthy fetus generally tolerates the stress of labor well. However, placental abnormalities, underlying maternal disease, or excessive uterine activity may cause a normal fetus to be adversely affected by the stress of labor. A fetus that is inherently compromised (e.g., growth restriction, viral infection, or a genetic disease) may not have the necessary reserve and may therefore exhibit an abnormal fetal heart rate pattern during labor. The term fetal distress has too often been applied to all major or minor variations in the fetal heart rate. Such usage has led to unwarranted conclusions as to fetal outcome, both immediate or long-term in relation to intrapartum fetal heart rate patterns.
The duration and extent of metabolic acidosis and hypoxia that will result in neurologic damage to the human fetus are not known. A specific umbilical artery value that defines acidemia and results in pathologic neonatal sequelae is not clear. The exact lower limit of newborn pH (metabolic acidemia) and depression (low 5-minute Apgar score) that are predictive of subsequent neurologic dysfunction have not been determined.
Umbilical Cord Acid-Base Determinants: Normal Values
Various investigators have established normal ranges for umbilical artery pH and blood gas values.12-15 Most have found that the mean umbilical artery pH ranges from 7.25 to 7.30 with 2 SD below the mean ranging from 7.15 to 7.20. Thorp et al.12 have reported a large series in which normal ranges for umbilical cord blood gas levels at term were established. They determined the mean umbilical artery pH to be 7.24 with SD of 0.07 pH units. Thus, in their series a pH of 7.10 was a statistically low pH, i.e., >2 SD below the mean umbilical artery pH. Ruth and Raivio16 selected an umbilical artery pH of <7.16 as >2 SD below the mean. We have previously reported that 2 SD below the mean umbilical artery pH is 7.18.13 However, the population from which this figure was derived was selected to have been at term gestation, to have had a spontaneous vaginal delivery and to have had a normal fetal heart rate baseline and variability in the second stage of labor. Thus, despite the traditional definition of umbilical artery acidemia as a pH <7.20,17 most recognize that this level is arbitrarily high and may range as low as the 7.10 suggested by Thorp et al.12
Applicable normal values for umbilical cord blood as reported by Yeomans et al.13 are shown in Table 1 and a suggested classification for type of fetal acidemia as reported by Gilstrap et al.18 in Table 2.
TABLE 1: Normal Values for Umbilical Cord Blood*
*Results are for 146 newborns after uncomplicated labor and vaginal delivery at 37 - 47 weeks of gestation. Values are mean ± standard deviation; ranges are given in parentheses.
| CORD BLOOD | pH | PCO2 (mmHG) |
PO2 (mmHG) |
BICARBONATE (meq/L) |
|---|---|---|---|---|
| Arterial | 7.28 ± 0.05 (7.15 - 7.43) |
49.2 ± 8.4 (3.11 - 74.3) |
18.0 ± 6.2 (3.8 - 33.8) |
22.3 ± 2.5 (13.3 - 27.5) |
| Venous | 7.35 ± 0.05 (7.24 - 7.49) |
38.2 ± 5.6 (23.2 - 49.2) |
29.2 ± 5.9 (15.4 - 48.2) |
20.4 ± 2.1 (15.9 - 24.7) |
TABLE 2: Classification of Fetal or Newborn Acidemia*
*Umbilical artery pH less than 7.20.
+ Means ± standard deviations are given in parentheses.
| ACIDEMIA TYPE | PCO2 (mmHG) |
HCO3¯ | BASE DEFICIT (meq/L)+ |
|---|---|---|---|
| Respiratory | High (>65) |
Normal (=22) |
Normal (6.4 ± 1.9) |
| Metabolic | Normal (<65) |
Low (=17) |
High (-15.9 ± 2.8) |
| Mixed | High (=65) |
Low (=17) |
High (-9.6 ± 2.5) |
Collection of Umbilical Cord Blood
Immediately after delivery of the neonate, a segment of umbilical cord should be doubly clamped, divided, and placed on the delivery table pending assignment of the 5-minute Apgar score. Although it is preferable to obtain umbilical arterial blood for analysis, there are situations, such as marked prematurity, in which venous blood is more readily sampled. Occasionally, it may be difficult to obtain either. A clamped segment of cord is stable for pH and blood gas assessment for at least 15 minutes, and a cord blood sample in a 1 ml syringe flushed with a heparin solution, 1,000 U/m1, is stable for up to 60 minutes.19 If the 5-minute Apgar score is satisfactory and the infant appears stable and vigorous, the segment of umbilical cord can be discarded. If any serious abnormality in the delivery process or problem with the neonate's condition persists at or beyond the first 5 minutes, blood can be drawn from the cord segments and sent for blood gas analysis.
Correlation of Umbilical Cord Gases with Subsequent Outcomes
Neonatal
No one has demonstrated any significant biologic correlation between the statistical normals and definitions in Tables 1 and 2 and the subsequent immediate neonatal performance. We have surmised, as did Gilstrap et al.20 and as supported by Low et al.21 that one explanation for the apparent lack of correlation of umbilical artery cord gas parameters and immediate neonatal or long-term asphyxic sequelae has been the utilization of one standard deviation below statistical norms of the umbilical artery pH as the abnormal baseline for comparison. The inclusion of newborns who have a statistically significant low pH value (with the reported lower cutoff values of 7.10, 7.15, 7.18 or 7.20) has had little correlation to fetal biologic damage at term gestation, as reflected by a newborn course compatible with intrapartum asphyxia. It is apparent that the biologic effect of an intrapartum period of substantial hypoxia that is sufficient to cause protracted fetal and neonatal damage is not a linear function.
Gilstrap et al.20 defined birth asphyxia in 2,738 term pregnancies that resulted in live births on the basis of umbilical cord pH, Apgar score and newborn cerebral dysfunction. Most (86 percent) had a Ua pH ³ 7.20 and only 18 newborns (0.6 percent) had a pH <7.00. Newborn complications did not increase in a linear fashion with a decreasing umbilical cord pH. Complications were significantly increased only below a pH of 7.00. (Tables 3 and 4). Seizures were more frequent in newborns with both an umbilical artery cord pH <7.00 and 1-minute Apgar £ 3 (Table 5). They concluded that newborns with a pH <7.00 and a 1-minute Apgar score £ 3 had increased morbidity (p<0.05). All required resuscitation and one half had neurologic dysfunction.
TABLE 3: Neonatal Complications at Term in Relation to U pH
| pH | ||
| =7.25 (N=1833) |
<7.00 (N=18) |
|
|---|---|---|
| Intubation | 13 | 6 |
| Respiratory | 45 | 5 |
| Hypotonia | 35 | 3 |
| 93 (5%) | 14 (78%) | |
TABLE 4: Neonatal Complications at Term in Relation to UapH
| % | |
|---|---|
|
>7.25 |
5 |
| >7.19 | 4 |
| >7.15 | 8 |
| >7.09 | 16 |
| >7.00 | 18 |
| <7.00 | 78 |
Winkler et al.22 correlated umbilical artery blood gas values in 2,764 term pregnancies with newborn morbidity. Similar to Gilstrap et al.20 13 percent had an Ua pH <7.20 and 0.8 percent were <7.00. The 358 newborns with a pH <7.20 were matched with a term delivery that occurred during the same time period, and with similar inclusion criteria except that the UapH was ³ 7.20. Only two newborns had neonatal complications ascribed to birth asphyxia. Both had a metabolic acidemia with an Ua pH of <7.00 and a 5-minute Apgar score of £ 3. Thus, 2/23 infants with an Ua pH <7.00 as compared to 0/793 of the remaining infants with a pH >7.00 had any neonatal complications ascribed to birth asphyxia (p<0.00l) as determined by attending neonatal faculty evaluation. Ramin and Gilstrap et al.23 confirmed their findings regarding an increase in neonatal complications with a pH <7.00 in 1,208 newborns with a birth weight of £ 2,500 g. Goldaber and Gilstrap et al.24 extended their observations regarding pathologic fetal acidemia. In 3,506 term newborns they found that 15 of 87 (17 percent) newborns with an umbilical pH of <7.00 had a neonatal death or unexplained seizure as compared to 11 of 3,419 (0.3 percent) newborns whose pH was >7.00 (Table 6).
Most recently, Goodwin and Paul et al.25 reviewed the neonatal outcome in 129 term nonanomalous singleton infants with an umbilical cord blood pH of <7.00. They concluded that infants with this degree of umbilical acidemia can be separated with regard to the risk of hypoxic-ischemic encephalopathy and abnormal neurologic outcome by consideration of the severity and composition of the acidemia and evidence of other end-organ dysfunction. Even in this pH range the Apgar score was not highly predictive of asphyxial complications.
TABLE 5: Newborn with Seizures in Relation to Umbilical Cord pH and 1-Minute Apgar Score
| Seizures | |
|---|---|
| pH <7.00 Apgar > 3 |
0/13 |
| pH >7.00 Apgar <3 |
2/49 (4%) |
| pH <7.00 Apgar <3 |
2/5 (40%) |
Long-term Follow-up
The infrequent association of newborn acidemia and subsequent cerebral palsy has been known for some time.26 More recently, this has been confirmed by Ruth and Raivio,16 Dijxhoorn et al.,5 Dennis et al.,27 and Fee et al.28 Ruth and Raivio16 confirmed cerebral palsy at 1 year of age in 4 of 982 infants. None of these four infants had an Ua pH less than the currently accepted lower limit of normalcy (7.15). Dijxhoorn et al.5 in a study of 805 term newborns reported a normal neurologic examination in 9 newborns whose Ua pH was <7.00 and that the 30 infants with abnormal neurologic findings had the same Ua pH distribution and mean as did the neurologically normal infants. These reports are consistent with Stanley's8 report that the incidence of cerebral palsy is 1-2/1,000 term infants. Thus, these reports confirm that severe metabolic acidemia, even if present, is infrequently associated with subsequent cerebral palsy.
TABLE 6: Frequency of Neonatal Deaths, All Seizures, and Unexplained Seizures in Term Infants According to Umbilical Blood pH Levels
| <7.00 (N=87) |
7.00-7.04 (N=95) |
7.05-7.09 (N=290) | 7.10-7.14 (N=798) | 7.15-7.19 (N=2236) | |
|---|---|---|---|---|---|
| Neonatal Deaths |
7 (8%) |
1 (%) | 0 | 3 (0.4%) | 3 (0.1%) |
| All seizures | 11 (12.6%) p=.06 |
4 (4.2%) p=.004 |
0 | 2 (0.3%) | 4 (0.2%) |
| Unexplained seizures | 8 (9.2%) p=.01 |
1 (1%) | 0 | 1 (0.1%) | 2 (0.1%) |
| Neonatal death and seizures | 2 (2.3%) | 1 (1%) | 0 | 0 | 1 (.05%) |
FIGURE 1: Major Impairment vs. Umbilical Bicarbonate
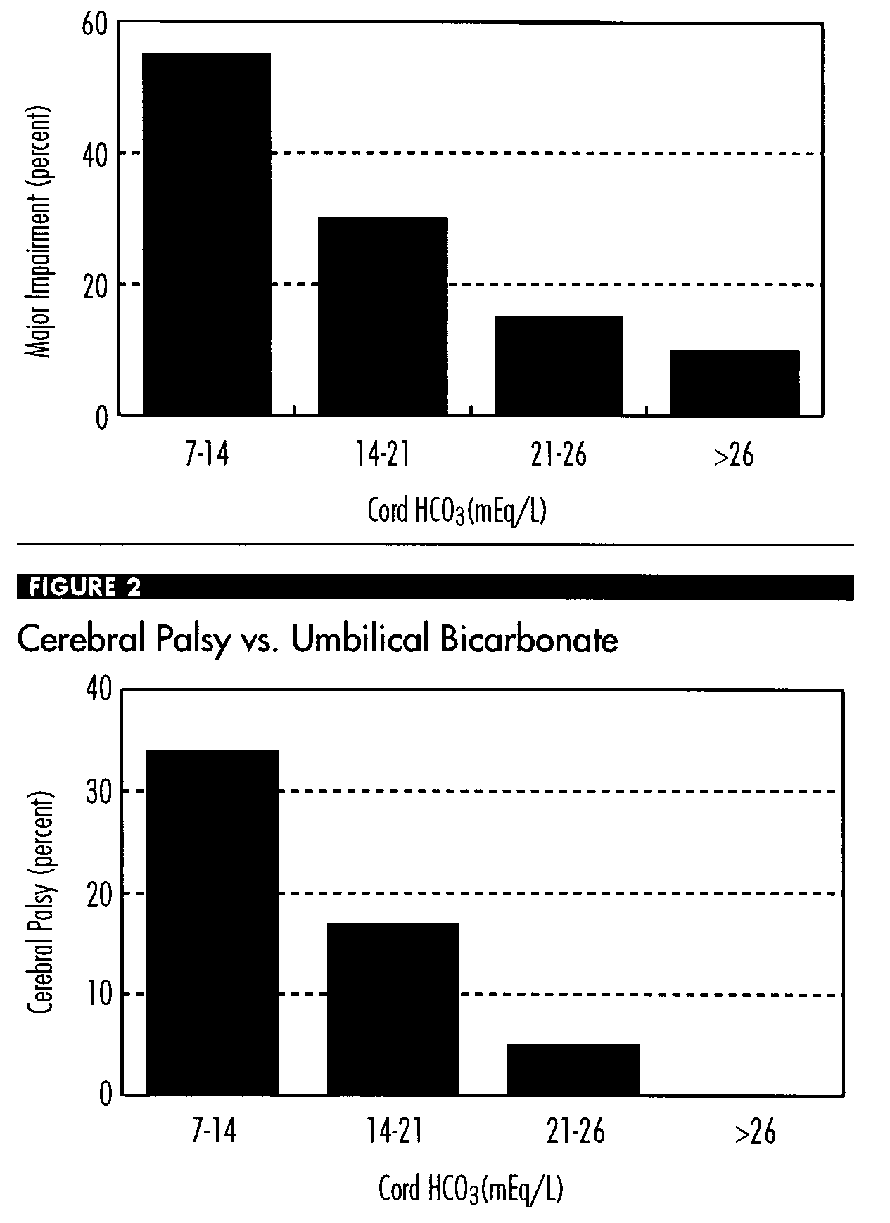
However, the precise association of severe newborn asphyxia, as defined as a metabolic acidemia with Ua pH of <7.00 and a 5- minute Apgar < 3, with neurologic status at one year of age is not known. These two reports16,5 included too few newborns with these findings to address this question.
Gaudier et al.29 has, however, provided evidence, albeit in markedly preterm newborns, that metabolic acidemia does increase the incidence of neurologic dysfunction at one year of age. They found that in 215 infants with a birth weight of <1,000 g there was a significant inverse correlation with major neurologic impairment and cerebral palsy at one year of age as compared to umbilical cord HCO3 (mEq/L) levels (Figures 1 and 2).
In summary, we believe that there is an increased incidence of newborn morbidity related to asphyxia in term newborns as defined as an umbilical artery pH of less than 7.0, a 5-minute Apgar score of 3 or less and in whom the acidemia has a metabolic component. In the depressed newborn, the documentation of umbilical blood acid-base measurements provides an objective fetal assessment and, if levels are within the normal range, can exclude intrapartum hypoxia as a proximate cause of neonatal depression.
Intrapartum Fetal Acid-Base Assessment
Fetal scalp capillary blood sampling was introduced by Saling in 1962. It can provide assessment of the fetal acid-base status. However, because of its limited availability, prerequisite for use, and invasive nature it has been estimated that fetal scalp blood sampling is used in less than 1 percent of all deliveries.17 Generally, this technique is used as an adjunct to fetal heart rate (FHR) monitoring. It is especially useful when a nonreassuring FHR pattern is encountered. It can be used as primary means of intrapartum assessment when variant heart rate patterns are present, such as fetal bradyarrhythmias.
Tejani et al.30 reported that FHR monitoring can be used to screen the innocuous from the ominous periodic changes but that fetal scalp blood pH must be obtained to identify accurately the true from false positive ominous patterns. They also found that a fetal scalp blood pH of £ 7.2 more accurately predicted neonatal depression than did FHR monitoring alone.31 Zalar and Quilligan32 reported that scalp pH sampling clarified the diagnosis of fetal distress and reduced the number of cesarean deliveries by approximately 10 percent. Young et al.33 also found that determining fetal blood pH avoided 10 percent of their cesarean deliveries. In 1985, from the same institution as Zalar and Quilligan, Clark and Paul34 concluded that "a properly trained clinician may pursue an approach for the detection of fetal distress that does not include scalp blood sampling without either compromising his ability to detect fetal distress or significantly increasing the cesarean section rate."
In May 1993, Goodwin and Paul et al.35 reviewed databases for scalp pH, cesarean delivery for fetal distress, Apgar score, neonatal diagnosis of asphyxia, and meconium aspiration syndrome for 6 years, 1986-1991. The yearly number of deliveries was 18,000 and the rate of scalp sampling for the first 3 years of the study period was 1.8 percent of all deliveries. That rate was consistent with the 1.5-2.0 percent noted for the preceding decade at their institution. The increase in 1987 was followed by a steady decline over the next 4 years (Figure 3). This decline coincided with the widespread application of intrapartum vibro-acoustic stimulation and the appreciation that the presence of fetal heart rate accelerations (spontaneous or provoked) precluded significant metabolic acidosis. During the period of declining scalp pH usage there was no significant change in the rate of cesarean delivery for fetal distress (2.5-3.0 percent), low Apgar score (less than 5 at 5 minutes), or clinical diagnosis of newborn asphyxia. Other measures of neonatal morbidity, including meconium aspiration syndrome, were unchanged. They noted that scalp Ph is currently used in less than 1/2,000 deliveries at their institution. Fetal scalp sampling has been virtually eliminated at this institution without an increase in the cesarean delivery rate for fetal distress or in the incidence of neonatal depression. They concluded that there is little, if any, place for fetal scalp sampling in contemporary obstetric practice.
FIGURE 3: Rates of Fetal Scalp Sampling at the University of Southern California (1986-1991)
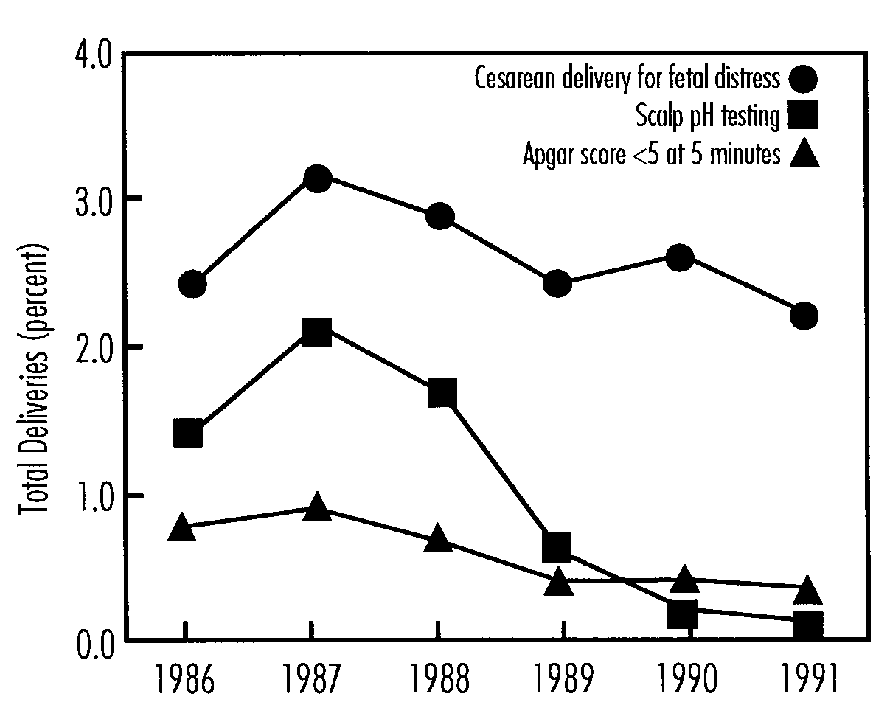
Fetal scalp blood sampling is performed via vaginoscopy. The cervix must be 2-3 cm dilated, the membranes must be ruptured, and the presenting part should be engaged. Fetal blood is collected in capillary tubes from a small scalp puncture and analyzed for pH and/or PO2, HCO3 and PCO2 with appropriately calibrated equipment. A fetal scalp pH of 7.25 or more is considered normal and is usually repeated based on progress in labor and changes in the fetal heart rate pattern. Values between 7.20 and 7.25 are considered borderline and are repeated at 15- to 30-minute intervals. Values below 7.20 are generally thought to be an indication for delivery.36
Research Needs
Data
- In term infants a prospective study of the correlation of the degree and type of Ua acidemia and or very low Apgar scores versus serious neonatal morbidity.
- In term infants a prospective follow-up to one year of age of all newborns with an Ua pH of £ 7.10 or a 5-minute Apgar score of £ 3 and as compared to matched control with an Ua, pH of £ 7.15 and 5-minute Apgar score of ³ 7.
Technology
- Development of a reliable, efficient and easy-to-utilize means to continuously measure fetal pH levels; or,
- Development of a means to easily assess anaerobic metabolism (presumably due to hypoxia) in the fetus, such as lactate or bicarbonate determinations.
References
- Little WJ. On the influence of abnormal parturition, difficult labors, premature birth, and asphyxia neonatorum, on the mental and physical condition of the child, especially in relation to deformities. Trans Obstet Soc (London) 2:293-344, 1862.
- US Department of Health and Human Services; Public Health Service; National Institutes of Health; National Institute of Child Health and Human Development; Freeman JM (ed): Prenatal and Perinatal Factors Associated with Brain Disorders, NIH Publication No. 85-1149. Washington DC, US Government Printing Office, 1985.
- Blair E, Stanley FJ. Intrapartum asphyxia: A rare cause of cerebral palsy. J Pediatr 112:515-519, 1988.
- Nelson KB, Ellenberg JH. Antecedents of cerebral palsy: Multivariate analysis of risks. N Engl J Med 315:81-86, 1986.
- Dijxhoorn MJ, Visser GHA, Fidler VJ et al. Apgar score, meconium and acidemia at birth in relation to neonatal neurologic morbidity in term infants. Br J Obstet Gynecol 93:217-222, 1986.
- Torfs CP, van den Berg BJ, Oechsli FW, Cummins S. Prenatal and perinatal factors in the etiology of cerebral palsy. J Pediatr 116:615-619, 1990.
- Melone PJ, Ernest JM, O'Shea MD, Klinepeter KL. Appropriateness of intrapartum fetal heart rate management and risk of cerebral palsy. Am J Obstet Gynecol 165:272-277, 1991.
- Stanley FJ, Watson L. The cerebral palsies in Western Australia: Trends, 1968 to 1981. Am J Obstet Gynecol 158:89-93, 1988.
- Committee on Obstetrics: Maternal-Fetal Medicine (ACOG) and Committee on Fetus and Newborn (AAP). Use and misuse of the Apgar score. Washington, DC: American College of Obstetricians and Gynecologists, ACOG Committee Opinion No. 49, November 1986.
- Nelson KB, Ellenberg JH. Apgar scores as predictors of chronic neurologic disability. Pediatr 68:36-44, 1981.
- Committee on Obstetrics: Maternal-Fetal Medicine (ACOG). Utility of umbilical cord blood acid-base assessment. Washington, DC: American College of Obstetricians and Gynecologists, ACOG Committee Opinion No. 91, February 1991.
- Thorp JA, Sampson JE, Parisi VM, Creasy RK. Routine umbilical cord gas determinations. Am J Obstet Gynecol 161:600-605, 1989.
- Yeomans ER, Hauth JC, Gilstrap LC III, Strickland DM. Umbilical cord pH, PCO2, and bicarbonate following uncomplicated term vaginal deliveries. Am J Obstet Gynecol 151:798-800, 1985.
- Wible JL, Petrie RH, Koons A, Perez A. The clinical use of umbilical cord acid-base determinations in perinatal surveillance and management. Clin Perinatol 9:387-397, 1982.
- Low JA, Pancham SR, Worthington D, Boston RW. Acid-base, lactate and pyruvate characteristics of the normal obstetrical patient and fetus during the intrapartum period. Am J Obstet Gynecol 120:862-867, 1974.
- Ruth VJ, Raivio KO. Perinatal brain damage: Predictive value of metabolic acidosis and the Apgar score. Br Med J 297:24-27, 1988.
- American College of Obstetricians and Gynecologists Technical Bulletin. Assessment of Fetal and Newborn Acid-Base Status, 127, 1989.
- Gilstrap LC, Hauth JC, Hankins GDV, Beck AW. Second-stage fetal heart rate abnormalities and type of neonatal acidemia. Obstet Gynecol 70:191-195,1987.
- Strickland DM, Gilstrap LC III, Hauth JC et al. Umbilical cord pH and PCO2: Efficient of interval from delivery to determination. Am J Obstet Gynecol 148:191-193, 1984.
- Gilstrap LC, Leveno JK, Burns J et al. Diagnosis of birth asphyxia on the basis of fetal pH, Apgar score, and newborn cerebral dysfunction. Am J Obstet Gynecol 161:825-830, 1989.
- Low JA, Galbraith RS, Muir DW et al. Intrapartum fetal hypoxia: A study of long-term morbidity Am J Obstet Gynecol 145:129-134, 1983.
- Winkler CL, Hauth JC, Tucker JM et al. Neonatal complications at term as related to the degree of umbilical artery acidemia. Am J Obstet Gynecol 164:637-641, 1991.
- Ramin SM, Gilstrap LC III, Leveno KJ et al. Umbilical artery acid-base status in the preterm infant. Obstet Gynecol 74:256-258, 1989.
- Goldaber KG, Gilstrap LC III, Leveno KJ et al. Pathologic fetal acidemia. Obstet Gynecol 78:1103-1107, 1991.
- Goodwin TM, Belai I, Hernandez et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol 162:1506-1512, 1992.
- Keith HM, Gage RP Neurologic lesions in relation to asphyxia of the newborn and factors of pregnancy: Long-term follow-up. Pediatrics 26:616 -622,1960.
- Dennis J, Johnson A, Mutch L et al. Acid-base status at birth and neurodevelopmental outcome at four and one-half years. Am J Obstet Gynecol 161:213-220, 1989.
- Fee SC, Malee K, Deddish R et al. Severe acidosis and subsequent neurologic status. Am J Obstet Gynecol 162:802-806, 1990.
- Gaudier FL, Goldenberg RL, Peralta M et al. Prediction of long term neurologic handicap in very low birthweight newborns. Am J Obstet Gynecol 166:419, 1992.
- Tejani N, Mann LI, Bhakthavathsalan A, Weiss RR. Correlation of fetal heart rate-uterine contraction patterns with fetal scalp blood pH. Obstet Gynecol 46:392-396, 1975.
- Tejani N, Mann LI, Bhakthavathsalan A. Correlation of fetal heart rate patterns and fetal pH with neonatal outcome. Obstet Gynecol 48:460- 463, 1976.
- Zalar RW, Quilligan EJ. The influence of scalp sampling on the cesarean section rate for fetal distress. Am J Obstet Gynecol 135:239-246, 1979.
- Young DC, Gray JH, Luther ER, Peddle LJ. Fetal scalp blood pH sampling: Its value in an active obstetric unit. Am J Obstet Gynecol 136:276- 281, 1980.
- Clark SL, Paul RH. Intrapartum fetal surveillance: The role of fetal scalp blood sampling. Am J Obstet Gynecol 153:717-720, 1985.
- Goodwin TM, Masterson LM, Vanis RW, Paul RH. What is the role of the fetal scalp pH in contemporary obstetric practice? American College of Obstetricians and Gynecologists Annual Meeting, Washington, DC, May 1993.
- Hon EH, Khazin AF. Biochemical studies of the fetus I. The fetal pH monitoring system. Obstet Gynecol 33:219, 1969.
Fetal Monitoring: Role of Doppler Blood Flow Velocity and Fetal Heart Rate in Assessment of Fetal Asphyxia
James A. Low, M.D.
Kingston General Hospital, Ontario, Canada
Introduction
Asphyxia is defined as a condition due to lack of oxygen in respired air resulting in impending or actual cessation of apparent life. Fetal asphyxia is asphyxia in utero due to anoxia.1 Increasing hypoxemia leads to fetal compromise due to tissue hypoxia, anaerobic metabolism and a metabolic acidosis. Tissue hypoxia of a particular degree and duration will cause multiple organ damage including the brain.
Confusion surrounds the term birth asphyxia.2 This is because the term has been used to include not only the exposure-response elements of asphyxia but also a component of the outcome reflected by permanent neurological impairment.
The objective of this review is to examine the relationship between a number of clinically observable signs, i.e., antepartum Doppler blood flow velocity and antepartum and intrapartum fetal heart rate to the exposure-response measures of fetal asphyxia and the outcome attributable to asphyxial brain damage.
Doppler Blood Flow Velocity
Prediction of Clinical Fetal Distress
A wide range of studies have addressed the predictive value of Doppler blood flow velocity for clinical fetal distress during labor. Study populations have included pregnancies at risk because of one or more maternal, obstetric, or fetal complication, or general obstetric populations. Maternal or fetal blood flow velocity observations have been obtained at varied times between 18 weeks and early labor. The outcome measures have usually been clinical fetal distress leading to intervention by either operative vaginal delivery or cesarean section. Diagnosis of fetal distress has been based on some combination of meconium in the amniotic fluid and abnormality of fetal heart rate, including decreased baseline variability or fetal heart rate decelerations.
Two studies have provided data on the association of uteroplacental blood flow velocity and fetal distress in labor. Schulman et al.3 in a study of 71 selected pregnancies observed a higher incidence of fetal distress in pregnancies with abnormal uteroplacental systolic/diastolic ratios (27 percent), in relation to pregnancies with normal systolic/diastolic ratios (5 percent). On the other hand, Brar et al.4 in a study of 92 selected pregnancies observed no association between uteroplacental systolic/diastolic ratios and fetal distress.
Four studies of selected risk pregnancies have provided data on the association between fetal blood flow velocity and fetal distress in labor. Three studies have demonstrated a significant association between absent end-diastolic velocity in the aorta5,6 and the umbilical artery7 and fetal distress in labor. Brar et al .8 presented data to indicate that measurement of umbilical artery blood flow velocity can identify late decelerations observed before or in early labor.
Two studies of growth retardation have examined the association between umbilical artery blood flow velocity and fetal distress in labor. Rochelson et al.9 in a study of 54 pregnancies observed fetal distress in 20 of 38 (53 percent) with abnormal umbilical artery systolic/diastolic ratios. Brar et al.10 in a study of eight pregnancies with reversed end-diastolic velocity observed fetal distress during labor in six of the eight cases (75 percent).
Four studies of post-term pregnancies have examined the association between uteroplacental and umbilical artery blood flow velocity and fetal distress in labor. Rightmire et al.11 observed a small increase in umbilical artery pulsatility index in the post-term fetus with fetal distress. However, three studies have found no evidence that uteroplacental12 or umbilical artery blood flow velocity12- 14 can identify the post-term fetus at risk of fetal distress during labor. These authors concluded that maternal or fetal blood flow velocity studies are unlikely to be useful in the assessment of fetal status in prolonged pregnancy.
Studies of selected-risk pregnancies have provided data on the predictive value of fetal blood flow velocity for fetal distress during labor (Table 1). Two studies have demonstrated that absent end-diastolic blood flow velocity in the fetal aorta15 and in the umbilical artery,16 may be useful predictors of fetal distress with a sensitivity of 83 percent and 75 percent, respectively. However, abnormal umbilical artery blood flow velocities, are not as effective predictors, with sensitivities ranging from 33 percent to 57 percent. The limitations of these observations are further emphasized by the low positive predictive values, ranging from 24 percent to 50 percent.16- 19
Random studies of general obstetric populations have provided data on the predictive value of maternal and fetal blood flow velocity for fetal distress during labor. Hanretty et al.20 examined 357 random pregnancies between 26 and 30 weeks gestation. Newnham et al.21 examined 535 random pregnancies at 18, 24, 28 and 34 weeks. Both studies concluded that uteroplacental blood flow velocity measures at these gestational ages were of little value in the prediction of clinical fetal distress in labor. Similarly, although there was some evidence of an association between abnormal umbilical artery blood flow velocity and clinical fetal distress during labor, both the sensitivity and positive predictive values are low.
Two studies of random obstetric patients have examined the relationship between umbilical artery blood flow velocity in early labor and clinical fetal distress in labor. Feinkind et al.22 observed an association between abnormal umbilical artery systolic/diastolic ratios and fetal distress. However, both the sensitivity and positive predictive values were low Sarno et al.23 in a smaller study, found no relationship between abnormal umbilical artery systolic/diastolic ratios in early labor and clinical fetal distress during labor.
TABLE 1: Predictive Value of Fetal Blood Flow Velocity for Fetal Distress in Labor
FA-fetal aorta; UA - umbilical artery; absent end-diastolic velocity; S/D - systolic to diastolic ratio.
*Selected pregnancies at risk.
+ Random population.
| Year | Series | Study Population (N) |
Fetal Distress (%) |
Protocol | Sensitivity (%) |
Specificity (%) |
Positive Predictive Value (%) |
Negative Predictive Value (%) |
|
|---|---|---|---|---|---|---|---|---|---|
| Site | Index | ||||||||
| 1987 | Laurin et al. 15 | 159* | 19 | FA | AEDV | 83 | 90 | 66 | 96 |
| 1987 | Rochelson et al. 16 | 161* | 13 | UA | S/D | 57 | 72 | 24 | 92 |
| 1988 | Berkowitz et al. 17 | 172* | 12 | UA | S/D | 43 | 81 | 24 | 91 |
| 1988 | Famakides et al. 18 | 140* | 27 | UA | S/D | 53 | 76 | 45 | 81 |
| 1990 | Newnham et al. 19 | 146* | 27 | UA | S/D | 33 | 88 | 50 | 78 |
| 1990 | Newnham et al. 21 | 469+
443+ |
9
9 |
UA
UA |
S/D
S/D |
12
15 |
95
95 |
18
23 |
91
92 |
| 1989 | Feinkind et al. 22 | 273+ | UA | S/D | 29 | 92 | 30 | 92 | |
TABLE 2: Correlation Between Doppler Measures of Blood Flow Velocity and Blood Gas Measures in Umbilical Vein Blood Obtained by Cordocentesis
Ao = aorta PI = pulsatility index *p <.05
UA = umbilical artery S/D = systolic to diastolic ratio **p<.01
IC = internal carotid BE = base excess ***p<.001
| Doppler Blood Flow Velocity | Blood Gas Measures | ||||||
|---|---|---|---|---|---|---|---|
| Study Population (N) |
Site Index | PO r |
PCO 2 r |
pH r |
BE r |
Lactate r |
|
| Soothill | 29 |
Ao Mean |
.73*** |
.54* |
.58** | .48* | |
| Ferrazzi | 9 |
UA PI |
|
.86** |
.98** |
.90** | |
| Weiner | 37 | UA S/D | .68*** | ||||
| Bilardo | 51 | UA PI |
.52 |
.40* |
.49* |
|
|
| Simonazzi | 17 | IC PI | .61* |
.68** |
|||
Association with Antepartum Blood Gas Measures
Cordocentesis has provided the opportunity to examine the relationship of Doppler blood flow velocity and umbilical vein blood gas measures in utero during the last half of pregnancy.
The vessels examined include the aorta, the umbilical artery and the cerebral circulation (Table 2).
Soothill et al.24 studied 29 suspected growth retarded pregnancies between 21 and 36 weeks gestational age. Mean blood flow velocity in the aorta was associated with hypoxemia, hypercapnia and acidosis.
Ferrazzi et al.25 examined the relationship between umbilical artery pulsatility index and umbilical vein blood gas measures in nine high-risk pregnancies sectioned between 30-35 weeks gestational age. Umbilical artery pulsatility index was not correlated with oxygen tension but was correlated with carbon dioxide tension. Increasing umbilical artery pulsatility index was associated with a decreasing pH and increasing lactate concentration. Weiner26 examined the relationship between umbilical artery systolic/diastolic ratio and umbilical vein blood gas measures in 165 pregnancies. There was no correlation before 25 weeks gestational age. However in fetuses after 25 weeks with a systolic/diastolic ratio greater than 3.5, there was a correlation between increasing systolic/diastolic ratios and decreasing oxygen tension. The six fetuses with persistent absent end-diastolic blood flow velocity were hypoxemic. Bilardo et al.24,27 examined the relationship between umbilical artery pulsatility index and umbilical vein blood gas measures in 51 pregnancies between 19 and 37 weeks gestational age. There was a correlation between umbilical artery pulsatility index and oxygen tension, carbon dioxide tension and pH.
The behavior of the cerebral circulation differs. Simonazzi et al.28 examined the relationship between internal carotid pulsatility index and scalp blood gases in 17 normal pregnancies. There was a significant correlation between pulsatility index and oxygen tension and base excess. The authors concluded that under physiological conditions cerebrovascular resistance was affected by changes in oxygen tension but not carbon dioxide tension. Bilardo et al.27 concluded that altered oxygen-carbon dioxide homeostasis was best represented by the inverse relationship of resistance in the aorta and common carotid artery. This is in keeping with the fetal response to hypoxia observed in the fetal lamb,29 i.e., an increase of blood supply to the brain with a reduction in the perfusion of the gastrointestinal tract, kidneys and lower extremities.
TABLE 3: Fetal Hypoxemia and Acidosis in Pregnancies with Abnormal or Absent End-Diastolic Blood Flow Velocity
EDV - end-diastolic velocity
| Umbilical Artery EDV | Total N | Hypoxemia N | Acidosis N |
|---|---|---|---|
| Nicolaides Absent | 59 | 47 | 27 |
| Nicolini Present | 26 | 12 | 8 |
| Nicolini Absent | 32 | 32 | 24 |
| Pardi Abnormal | 35 | 10 | 8 |
| Pardi Absent | 16 | 7 | 4 |
Several studies have examined the association between abnormal end-diastolic blood flow velocity and fetal blood gas characteristics (Table 3). Nicolaides et al.30 observed hypoxemia in 80 percent and acidosis in 46 percent of 59 pregnancies with absent end-diastolic blood flow velocity. Nicolini et al.31 reported on 58 pregnancies of whom 32 had absent end-diastolic blood flow velocity. Hypoxemia was present in 80 percent of those with no end-diastolic blood flow velocity and 45 percent of those with end-diastolic blood flow velocity. Pardi et al32 reported on 56 growth retarded pregnancies. Thirty-five had abnormal umbilical artery blood flow velocity of whom 16 had absent end-diastolic blood flow velocity. The majority of the abnormal blood gas measures, hypoxemia 44 percent and acidosis 25 percent occurred in the fetuses with absent end-diastolic blood flow velocity.
Abnormal fetal blood gases are a frequent occurrence particularly in the fetus with absent end-diastolic blood flow velocity. However, the presence of normal blood gases supports the contention that abnormal umbilical artery end-diastolic blood flow velocity precedes evidence of abnormal fetal heart rate or fetal hypoxemia and acidosis.33
Prediction of Fetal Blood Gas Measures at Delivery
Some evidence of a relationship between fetal blood flow velocity and umbilical vein and artery blood gases at delivery have been reported. Laurin et al.34 in a study of 159 pregnancies at risk for growth retardation calculated that when the pulsatility index in the aorta had a sensitivity of 90 percent for an umbilical vein pH < 7.2, that the false positive 24 rate was 50 percent. Ferrazzi et al.25 in a study of 14 high-risk pregnancies delivered by section between 30 and 35 weeks gestational age reported a significant correlation between umbilical artery pulsatility index and umbilical artery P02 (r 0.72), pH (r 0.86), PCO2 (r 0.89), base excess (r 0.70) and lactate (r 0.91).
Two studies have reported a relationship between absent end-diastolic blood flow velocity and fetal blood gases at delivery. Brar et al.10 in a study of pregnancies with a reversal of umbilical artery blood flow velocity observed an umbilical artery pH <7.20 in four of eight fetuses. Tyrrell et al.35 reported on 112 pregnancies with Doppler blood flow velocity studies within 4 hours of an elective section. There were 17 patients with absent end-diastolic blood flow velocity. Absent end-diastolic blood flow velocity was associated with hypoxemia (sensitivity 78 percent, positive predictive value 88 percent) and acidosis (sensitivity 90 percent, positive predictive value 53 percent).
However these associations have not been consistent in all studies. McCowan et al.36 in a study of 15 preterm growth retarded pregnancies observed no association between umbilical artery pulsatility index and umbilical artery pH at delivery. Divon et al.37 reported a similar umbilical artery pH in a matched cohort of fetuses with normal and abnormal umbilical artery systolic/diastolic ratios. Lowery et al.19 in a study of 146 risk pregnancies observed the same incidence of low pH in fetuses with normal and abnormal umbilical artery systolic/diastolic ratios.
Prediction of Reproductive Outcome
Preliminary evidence would indicate that abnormal umbilical artery blood flow velocity is predictive of a less favourable reproductive outcome. In a study of 58 growth retarded pregnancies, all 15 perinatal deaths occurred in those fetuses with absent end-diastolic blood flow velocity.31 In a study of pregnancies with abnormal fetal growth, oligohydramnios and non-reactive cardiotocographs, the prevalence of neonatal morbidity was significantly higher in fetuses with abnormal umbilical artery pulsatility index.38 A prospective follow-up study of 58 infants with absent end-diastolic blood flow velocity to 18 months corrected age demonstrated that 49 (83 percent) were neurologically normal while 9 (15 percent) were neurologically suspect or abnormal.40
Antepartum Fetal Heart Rate
Prediction of Fetal Outcome
Antepartum fetal heart rate assessment has become a widely accepted method of fetal surveillance. However, standardization of the test and its interpretation has not been achieved. Studies of the predictive value of the Non-Stress Test have included a range of outcome measures such as fetal growth retardation, fetal distress during labor, fetal acidosis at delivery, low Apgar scores, and fetal and neonatal deaths. A meta- analysis of the Non-Stress Test in relation to these outcome measures reported a sensitivity, 48 percent, specificity, 93 percent, positive predictive value, 38 percent, and negative predictive value, 95 percent.40 It is frequently stated that a reactive Non-Stress Test has a good negative predictive value. However, fetal deaths with a reactive Non-Stress Test are reported in many studies.41 Four randomized clinical trials have failed to demonstrate improved outcome with an antepartum Non-Stress Tests as an intermittent supplementary test of fetal well being in high-risk pregnancies.42-45
The biophysical profile adds observations of fetal breathing, movement and tone to amniotic fluid characteristics and fetal heart rate measures. An early report suggested this could be a good test of fetal well-being with only one unpredicted fetal death, a false negative rate of 0.8/1000.46 The predictive value of the biophysical profile for a wide range of outcome measures has reported a sensitivity 86 percent, specificity 89 percent, positive predictive value 75 percent, and a negative predictive value of 93 percent.47
Association With Antepartum Blood Gas Measures
The relationship between fetal heart rate and antepartum umbilical vein blood gas measures obtained by cordocentesis has been examined. This relationship was examined in 29 appropriate-for-gestational-age and 58 small-for-gestational-age fetuses between 27 and 38 weeks gestational age.48 The appropriate-for-gestational-age fetuses had blood gases in the normal range, while 27 of the 29 had a reactive fetal heart rate. Nineteen of the 58 small-for-gestational-age fetuses were hypoxemic, acidemic or both, of whom 15 had abnormal fetal heart rate patterns. The predictive values of fetal heart rate characteristics for hypoxemia and/or acidemia are summarized in Table 4. Absence of baseline variability and repetitive decelerations were the best predictors of hypoxemia and acidemia.
TABLE 4: The Negative and Positive Predictive Values of Fetal Heart Rate Characteristics for Hypoxemia and Acidemia
| Fetal Heart Rate | Blood Gas Measures | |
|---|---|---|
| Normal | Hypoxia and/or Acidemia | |
| Accelerations Present | 41 (85%) | 7 |
| Accelerations Absent | 7 | 12 (63%) |
| Baseline Variation >5 beats/minute | 47 (81%) | 11 |
| Baseline Variation <5 beats/minute | 1 | 8 (89%) |
| Decelerations Absent | 44 (88%) | 6 |
| Decelerations Present | 4 | 13 (76%) |
From: Visser et al.48
The relationship between computerized fetal heart rate recordings and umbilical vein blood gas measures has been examined in 25 growth retarded fetuses between 28 and 39 weeks gestational age.49 There was a significant correlation between fetal heart rate variability and oxygen tension (r=0.66) and pH (r=0.69). Both oxygen tension and pH were lower in the absence of accelerations and in the presence of decelerations. Long-term fetal heart rate variation less than 20 milliseconds was always associated with hypoxemia and acidemia. Fetal heart rate variability greater than 30 milliseconds without decelerations were usually normal. However, fetal heart rate variation between 20 and 30 milliseconds was associated with a large scatter of oxygen tension and pH values.
The relationship between the biophysical profile and umbilical vein blood gas measures was examined in 14 growth retarded fetuses.50 The biophysical profile score was correlated with pH (r=0.84) but not with PO2 or PCO2. The elements of the biophysical score with the best association with acidemia were oligohydramnios, absent fetal body movements and absent fetal tone.
Association With Blood Gas Measures at Delivery
Studies have examined the relationship between antepartum fetal heart rate and fetal blood gas measures at delivery in a number of different populations.
Several studies have demonstrated no association with acidosis but an association with hypoxemia. Low et al.51 examined the relationship between antepartum fetal heart rate patterns and fetal blood gas measures at delivery in 290 term fetuses delivered following an elective induction of labor. There was no correlation between fetal heart rate characteristics and metabolic acidosis at delivery. Bekedam et al.52 examined the relationship between antepartum fetal heart rate and fetal blood gases at delivery in 37 growth retarded fetuses delivered by elective section. The record of 29 fetuses had evidence of late decelerations. There was an association between late decelerations and low oxygen tension values in the umbilical vein and artery. There was no association with either respiratory or metabolic acidosis. Smith et al.53 examined the relationship with blood gas measures at delivery in 21 cases with abnormal fetal heart rate patterns delivered by elective section. There was an association between decreased heart rate variability and hypoxemia but no association with metabolic acidosis.
Two studies have indicated an association between antepartum fetal heart rate and acidosis. Vintzileos et al.54 examined the relationship between the fetal biophysical profile score and umbilical artery pH in 124 patients delivered by elective section. The predictive value of a biophysical profile score <7 for an umbilical artery pH <7.20 had a sensitivity, 90 percent and a positive predictive value, 82 percent. Dawes et al.55 examined the relationship between computerized antepartum measures of short-term fetal heart rate variations and decelerations as predictors of outcome in 3,563 patients.
TABLE 5: Relationship Between Blood Flow Velocity and Fetal Heart Rate and Fetal Asphyxia and Outcome
| ANTEPARTUM Hypoxemia & Acidosis |
0 | + | + | |
|---|---|---|---|---|
| INTRAPARTUM Clinical Fetal Distress |
0 | + | + | |
| DELIVERY Hypoxemia & Acidosis |
+/- | +/- | + | |
| NEWBORN Neurological Impairment |
? | + |
When short term variation exceeded 3 milliseconds, there were no intrauterine deaths and only one instance of metabolic acidosis at delivery. Fetal heart rate variation was 3 milliseconds or less in 98 patients. Thirty four percent of these subjects had either a metabolic acidosis at delivery or an intrauterine death.
Intrapartum Fetal Heart Rate
Electronic fetal heart rate monitoring was introduced into obstetric practice as a screening test for fetal asphyxia. The objective of this surveillance was a reduction of fetal and early neonatal mortality and fetal brain damage with its sequelae.56
In spite of the widespread application of this technology, there has been no evidence of a reduction of cerebral palsy in the term infant.57 However, its test characteristics were not well established prior to its widespread adoption.
Reliability
Fetal heart rate monitoring is a non-quantitative test dependent on pattern recognition. Thus intra-observer and inter-observer reliability may be a problem. This important question received relatively limited attention in the initial years of these programs. In recent years, a number of publications have described studies of inter-observer variability of the interpretation of fetal heart rate strips and whether they require intervention. The inter-observer reliability ranged from fair to poor (average Kappa value 0.4) in five such studies. Intra-observer reliability ranged from fair to good (average Kappa value 0.7), with remarkable divergences of interpretation in some cases.58 Such poor agreement may seriously obscure significant relationships. This problem may remain until computerization of fetal heart rate patterns during labor has been achieved.59,60
Validity
Laboratory and clinical studies have demonstrated an association between abnormal intrapartum fetal heart rate patterns and biochemically determined asphyxia. For example, in a matched case controlled study, there was an increased incidence of atypical variable or late decelerations in those fetuses with metabolic acidosis at delivery.61 The problem is that sensitivity of such decelerations is of the order of 50-60 percent, and the positive predictive value is of the order of 50 percent. The false positive rate can result in unnecessary intervention.62 Recent evidence suggests that the false positive rate can be offset in part by an ST waveform analysis of a concurrent electrocardiogram.63However, fetal scalp blood acid-base assessment is the appropriate diagnostic test in these circumstances. This practice will identify the false positives and may improve the sensitivity by extending the definition of what constitutes an abnormal fetal heart rate pattern.
Rosen and Dickinson64 reviewed a number of recent studies which examined the association between fetal heart rate patterns and neurologic morbidity. An association was observed between fetal heart rate patterns and Grade III or IV intracranial hemorrhage65,66 with neonatal seizures67,68 and neurologic abnormality.69-71 Collectively the sensitivity for an adverse outcome in these studies ranged from 23 percent to 100 percent, and the specificities from 45 percent to 100 percent. These studies emphasized the inconsistency of the classification of abnormal patterns of fetal heart rate. The authors concluded on the basis of this review that there is not a specific pattern of fetal heart rate predictive of neurologic abnormality.
Lumley72 reviewed studies that examined the relationship between fetal heart rate patterns and late neurodevelopmental outcome. Interpretation of these studies was difficult. There was no consistency of the fetal heart rate abnormalities used. The study populations varied, the duration of the follow-up and the method of assessment differed. However within these constraints, abnormal fetal heart rate patterns did show a modest correlation with later outcome. The probability of an abnormal outcome increased twofold with abnormal fetal heart rate patterns. Since the abnormal fetal heart rate patterns are common and neurologic abnormalities uncommon, the vast majority of infants with abnormal fetal heart rate patterns developed normally.
Prevention of Brain Damage
The ability of a test to predict a serious adverse outcome does not necessarily mean that clinical action taken on the basis of that test will prevent that outcome. A randomized trial of electronic fetal heart rate monitoring is required to determine if such intervention can prevent brain damage. Since the frequency of brain damage that can currently be identified is low, such randomized trials must be large.
Chalmers73 reported a meta-analysis of four randomized clinical trials representing a total of 2,032 patients which had been published at that time. Electronic fetal heart rate monitoring was associated with an increased section rate but this was modified if the protocol included fetal scalp blood sampling. Electronic fetal heart rate monitoring particularly with fetal scalp blood sampling decreased the incidence of neonatal seizures. However, there was no benefit in regard to abnormal neurological findings. The Dublin trial of intrapartum fetal monitoring randomly assigned 13,048 patients into two arms both of which included fetal scalp sampling. Electronic fetal monitoring was not associated with a decrease of mortality However there was a reduced risk for neonatal seizures.74 A follow-up assessment at age 4 demonstrated no difference in the incidence of cerebral palsy.75 These conclusions continue to be supported by ongoing clinical trials.77
Summary
In selected pregnancies, Doppler blood flow velocity and fetal heart rate are of predictive value for fetal asphyxia but have limited predictive value for asphyxial-induced brain damage.
There is a modest association between abnormal umbilical artery blood flow velocity particularly with absent end-diastolic blood flow velocity and intrapartum clinical fetal distress. These observations have predictive value with moderate sensitivity and low-to-moderate positive predictive value in selected pregnancies. However, they do not have a role as a screening test for clinical fetal distress in the general obstetric population.
There is evidence that decreased umbilical artery end-diastolic blood flow velocity reflects increasing placental vascular resistance in the fetal circulation due to placental pathology.77,78 Cordocentesis studies in growth retarded pregnancies have demonstrated an association between abnormal umbilical artery blood flow velocity and hypoxemia, hypercapnia and metabolic acidosis. The association between antepartum umbilical artery blood flow velocity and fetal blood gas measures at delivery has not been as consistent. Several studies have reported an association particularly between absent end-diastolic blood flow velocity and hypoxemia and acidosis at delivery. However, several studies have reported no association between abnormal umbilical artery blood flow velocity and acidosis at delivery.
These findings support the contention that abnormal umbilical artery blood flow velocity reflects placental pathology which if sufficiently severe will compromise blood gas exchange leading to fetal hypoxemia and in some cases metabolic acidosis. There is preliminary evidence that these findings may be associated with increased mortality, neonatal morbidity and long term neurological disability.
An abnormal Non-Stress Test and a low biophysical profile score are associated with abnormalities of reproductive outcome. However, randomized clinical trials have failed to demonstrate an improved reproductive outcome with intermittent antepartum Non-Stress Tests in high-risk pregnancies. Cordocentesis studies in growth retarded pregnancies have demonstrated an association between decreased heart rate variability and fetal heart rate decelerations and antepartum fetal hypoxemia and acidosis. Abnormal antepartum fetal heart rate characteristics may be associated with fetal hypoxemia at delivery. Metabolic acidosis at delivery has been reported following marked reduction of fetal heart rate variability.
These findings support the concept that the fetal cardiovascular response to hypoxemia includes reduced heart rate variability and heart rate deceleration as well as decreased fetal body movement and fetal breathing.
There are major problems in regard to the reliability of the interpretation of visually read intrapartum fetal heart rate recordings. These difficulties may not be resolved until computerized assessment of intrapartum fetal heart rate records has been achieved to standardize the interpretation of fetal heart rate patterns. In spite of these limitations there is an association between abnormal intrapartum fetal heart rate patterns and fetal hypoxemia and metabolic acidosis at delivery.
The diagnosis of fetal asphyxia requires a blood gas and acid-base assessment. There is increasing evidence that intrapartum fetal heart rate monitoring should be used as a diagnostic test in conjunction with appropriately scheduled fetal blood gas and acid base assessments. Randomized clinical trials indicate that such a protocol can reduce the incidence of neonatal seizures in the term newborn.
These tests of fetal blood flow velocity and fetal heart rate can be of predictive value for antepartum or intrapartum fetal hypoxemia or acidosis in selected pregnancies. Fortunately, the prevalence of fetal asphyxia and brain damage due to such asphyxial insults is low. Thus it will require large randomized clinical trials to determine that protocols incorporating these assessment measures will change the incidence of asphyxia or the organ system injuries associated with severe asphyxial insults.
References
- Dorland's Illustrated Medical Dictionary. 27th ed. Philadelphia: W.B. Saunders Co.
- Blair E. A research definition for "Birth Asphyxia." Develop Med Child Neurol 35:449-455, 1993.
- Schulman H, Ducey J, Farmakides G et al. Uterine artery Doppler velocimetry: The significance of divergent systolic/diastolic ratios. Am J Obstet Gynecol 157:1539-1542, 1987.
- Brar HS, Medearis AL, De Vore GR, Platt LD. Maternal and fetal blood flow velocity waveforms in patients with preterm labor: Relationship to outcome. Am J Obstet Gynecol 1611:1519-1522, 1989.
- Laurin J, Lingman G, Marsal K, Persson PH. Fetal blood flow in pregnancies complicated by intrauterine growth retardation. Obstet Gynecol 69:895-902, 1987.
- Lingman G, Laurin J, Marsal K. Circulatory changes in fetuses with imminent asphyxia. Biol Neonate 49:66-73, 1986.
- Reuwer PJ, Sijmons EA, Rietman GW et al. Intrauterine growth retardation: Prediction of perinatal distress by Doppler ultrasound. Lancet 2:415-418, 1987.
- Brar HS, Platt LD, Paul RH. Fetal umbilical blood flow velocity waveforms using Doppler ultrasonography in patients with late decelerations. Obstet Gynecol 73:363-366, 1989.
- Rochelson BL, Schulman H, Fleischer A et al. The clinical significance of Doppler umbilical artery velocimetry in the small-for-gestational- age fetus. Am J Obstet Gynecol 156:1223-1226, 1987.
- Brar HS, Platt LD. Reverse end-diastolic flow velocity on umbilical artery velocimetry in high-risk pregnancies: An ominous finding with adverse pregnancy outcome. Am J Obstet Gynecol 159:559-561, 1988.
- Rightmire DA, Campbell S. Fetal and maternal Doppler blood flow parameters in post-term pregnancies. Obstet Gynecol 69:891-894, 1987.
- Brar HS, Horenstein J, Medearis AL et al. Cerebral, umbilical, and uterine resistance using Doppler velocimetry in post-term pregnancy. J Ultrasound Med 8:187-191, 1989.
- Guidetti DA, Divon MY, Cavalieri RL et al. Fetal umbilical artery flow velocimetry in post-date pregnancies. Am J Obstet Gynecol 157:1521- 1523, 1987.
- Farmakides G, Schulman H, Ducey J et al.Uterine and umbilical artery Doppler velocimetry in post-term pregnancy. J Reprod Med 33:259-261, 1988.
- Laurin J, Marsal K, Persson PH, Lingman G. Ultrasound measurement of fetal blood flow in predicting fetal outcome. Br J Obstet Gynaecol 194:940-948, 1987.
- Rochelson B, Schulman H, Farmakides G et al. The significance of absent end-diastolic velocity in umbilical artery velocity waveforms. Am J Obstet Gynecol 156:1213-1218, 1987.
- Berkowitz GS, Mehalek KE, Chitkara U et al. Doppler umbilical velocimetry in the prediction of adverse outcome in pregnancies at risk for intrauterine growth retardation. Obstet Gynecol 71:742-746, 1988.
- Farmakides G, Schulman H, Winter D et al. Prenatal surveillance using non-stress testing and Doppler velocimetry. Obstet Gynecol 71:184-187, 1988.
- Lowery CL, Henson BV, Wan J, Brumfield CG. A comparison between umbilical artery velocimetry and standard antepartum surveillance in hospitalized high-risk patients. Am J Obstet Gynecol 162:710-714, 1990.
- Hanretty KP, Primrose MH, Neilson JP, Whittle MJ. Pregnancy screening by Doppler uteroplacental and umbilical artery waveforms. Br J Obstet Gynaecol 96:1163-1167, 1989.
- Newnham JP, Patterson LL, James IR et al. An evaluation of the efficacy of Doppler flow velocity waveform analysis; a screening test in pregnancy. Am J Obstet Gynecol 162:403-410, 1990.
- Feinkind L, Abulafia O, Delke I et al. Screening with Doppler velocimetry in labor. Am J Obstet Gynecol 161:765-770, 1989.
- Sarno AP, Ahu MO, Brar HS et al. Intrapartum Doppler velocimetry, amniotic fluid volume, and fetal heart rate as predictors of subsequent fetal distress. Am J Obstet Gynecol 161:1508-1514, 1989.
- Soothill PW, Nicolaides KH, Bilardo CM, Campbell S. Relationship of fetal hypoxia in growth retardation to mean blood velocity in the fetal aorta. Lancet 8516:1118-1120, 1986.
- Ferrazzi E, Pardi G, Bauscaglia M et al. The correlation of biochemical monitoring versus umbilical flow velocity measurements of the human fetus. Am J Obstet Gynecol 159:1081-1087, 1988.
- Weiner CP. The relationship between the umbilical artery systolic/diastolic ratio and umbilical blood gas measurements in specimens obtained by cordocentesis. Am J Obstet Gynecol 162:1198-1202, 1990.
- Bilardo CM, Nicolaides KH, Campbell S. Doppler measurements of fetal and uteroplacental circulations: Relationship with umbilical venous blood gases measured at cordocentesis. Am J Obstet Gynecol 162:145-120, 1990.
- Simonazzi E, Wladimiroff JW, van Eyck J. Flow velocity waveforms in the fetal internal carotid artery relative to fetal blood gas and acid- base measurements in normal pregnancy. Early Hum Dev 19:111-115, 1989.
- Peeters LLH, Sheldon RE, Jones MD, Jr et al. Blood flow to fetal organs as a function of arterial oxygen content. Am J Obstet Gynecol 135:637-646, 1979.
- Nicolaides KH, Bilardo CM, Soothill PW, Campbell S. Absence of end-diastolic frequencies in umbilical artery: A sign of fetal hypoxia and acidosis. Br Med J 297:1026-1027, 1988.
- Nicolini U, Nicolaides P, Fisk NM et al. Limited role of fetal blood sampling in prediction of outcome in intrauterine growth retardation. Lancet 336:768-72, 1990.
- Pardi G, Cetin I, Marconi AM et al. Diagnostic value of blood sampling in fetuses with growth retardation. N Engl J Med 328:692-696, 1993.
- Bekdedam DJ, Visser GHA, van der Zee et al. Abnormal velocity waveforms of the umbilical artery in growth retarded fetuses: Relationship to antepartum late heart rate decelerations and outcome. Early Hum Dev 79-89, 1990.
- Laurin J, Marsal K, Persson PH, Lingman G. Ultrasound measurement of fetal blood flow in predicting fetal outcome. Br J Obstet Gynaecol 94:940-948, 1987.
- Tyrrell S, Obaid AH, Lilford RJ. Umbilical artery Doppler velocimetry as a predictor of fetal hypoxia and acidosis at birth. Obstet Gynecol 74:332, 1989.
- McCowan LM, Erskine LA, Ritchie K. Umbilical artery Doppler blood flow studies in the preterm, small-for-gestational-age fetus. Am J Obstet Gynecol 156:655-659, 1987.
- Divon MY, Girz BA, Lieblich R, Langer O. Clinical management of the fetus with markedly diminished umbilical artery end-diastolic flow. Am J Obstet Gynecol 161:1523-1527, 1989.
- Ferrazzi E, Vegni C, Bellotti M et al. Role of umbilical Doppler velocimetry in the biophysical assessment of the growth retarded fetus. J Ultrasound Med 10:309-315, 1991.
- Kelly EN, Ryan G, Inwood S et al. Absent umbilical artery end diastolic velocity: Effects on long-term growth and development, Abstract S88. Ottawa: S.O.G. C., 1993.
- Devoe LD, Castillo RA, Sherline DM. The non-stress test as a diagnostic test: A critical reappraisal. Am J Obstet Gynecol 152:1047-1053, 1985.
- Phelan JP, Cromartie AD, Smith CV. The non-stress test. The false negative test. Am J Obstet Gynecol 142:293, 1982.
- Flynn AM, Kelly J, Mansfield H et al. A randomized trial of non-stress antepartum cardiotocography. Br J Obstet Gynaecol 89:427-433, 1982.
- Brown VA, Sawers RS, Parsons RJ et al. The value of antenatal cardiotocography in the management of high-risk pregnancy: A randomized control trial. Br J Obstet Gynaecol 89:716-722, 1982.
- Lumley J, Lester A, Anderrson I et al. A randomized trial of weekly cardiotocography in high-risk obstetric patients. Br J Obstet Gynaecol 90:1018-1026, 1983.
- Kidd LC, Patel NB, Smith R. Non-stress antenatal cardiotocography—a prospective randomized clinical trial. Br J Obstet Gynaecol 92:1156-1159, 1985.
- Manning FA, Baskett TE, Morrison I, Lange I. Fetal biophysical profile scoring: A prospective study in 1184 high-risk patients. Am J Obstet Gynecol 140:289, 1981.
- Devoe LD, Castillo RA, Searle N, Searle JS. Prognostic component of computerized fetal biophysical testing. Am J Obstet Gynecol 158:1148, 1988.
- Visser GHA, Sadowsky G, Nicolaides KH. Antepartum heart rate patterns in small for gestational age third-trimester fetuses: Correlations with blood gas values obtained at cordocentesis. Am J Obstet Gynecol 162:698-703, 1990.
- Ribbert LSM, Snijders RJM, Nicolaides KH, Visser GHA. Relation of fetal blood gases and data from computer-assisted analysis of fetal heart rate patterns in small-for-gestation fetuses. Br J Obstet Gynaecol 98:820-823, 1991.
- Ribbert LSM, Snijders RJM, Nicolaides KH, Visser GHA. Relationship of fetal biophysical profile and blood gas values at cordocentesis in severely growth retarded fetuses. Am J Obstet Gynecol 163:569-571, 1990.
- Low JA, McGrath MJ, Marshall SJ et al. The relationship between antepartum fetal heart rate, intrapartum fetal heart rate, and fetal acid- base status. Am J Obstet Gynecol 154:769-776, 1986.
- Bekadam DJ, Visser GHA, Mulder EJH, Poelmann-Weesjes G. Heart rate variation and movement incidence in growth retarded fetuses: The significance of antenatal late heart rate decelerations. Am J Obstet Gynecol 157:126-133, 1987.
- Smith JH, Anand KJS, Cotes PM et al. Antenatal fetal heart rate variation in relation to the respiratory and metabolic status of the compromised human fetus. Br J Obstet Gynaecol 95:980-989, 1988.
- Vintzileos AM, Gaffney SE, Salinger LM et al. The relationship among the fetal biophysical profile, umbilical cord pH and Apgar scores. Am J Obstet Gynecol 157:627-631, 1987.
- Dawes GS, Moulden M, Redman CWG. Short term fetal heart rate variation, decelerations, and umbilical flow velocity waveforms before labor. Obstet Gynecol 80:673-678, 1992.
- Quilligen EJ, Paul RH. Fetal monitoring: Is it worth it? Obstet Gynecol 45:96-100, 1975.
- Pharoah DD, Tooke T, Tooke RWI, Rosenbloom L. Birthweight specific trends in cerebral palsy Arch Dis Child 65:602-606, 1990.
- Paneth N, Bonmarito M, Stricker J. Electronic fetal monitoring and later outcome. Clin Invest Med 16:159-165, 1993.
- Nielsen PV, Stigsby B, Nickelson C. Computer assessment of the intrapartum cardiotocogram. Act Obstet Gynecol Scand 67:455-464, 1988.
- Pello C. Computerized fetal heart rate analysis in labor. Obstet Gynecol 78:602-610, 1991.
- Low JA, Cox MJ, Karchmar EJ et al. The prediction of intrapartum fetal metabolic acidosis by fetal heart rate monitoring. Am J Obstet Gynecol 139:299-305, 1981.
- Freeman R. Intrapartum fetal monitoring—a disappointing story. N Engl J Med 322:6214-6216, 1990.
- Westgate J, Harris M, Curnow JSH, Greene KR. Randomized trial of cardiotocography alone or with ST waveform analysis for intrapartum monitoring. Lancet 2:194-198, 1992.
- Rosen MG, Dickinson JC. The paradox of electronic fetal monitoring; More data may not enable us to predict or prevent infant neurologic morbidity. Am J Obstet Gynecol 168:745-751, 1993.
- Strauss A, Kirz D, Modanlou HD, Freeman RK. Perinatal events and intraventricular subependymal hemorrhage in the very-low-birth weight infant. Am J Obstet Gynecol 151:1022-1027, 1985.
- Westgren IM, Malcus P, Svenningsen NW. Intrauterine asphyxia and long-term outcome in preterm fetuses. Obstet Gynecol 67:512-516, 1986.
- Keegan KA, Wofforn F, Quilligan EJ. Obstetric characteritistics and fetal heart rate patterns of infants who convulse during the newborn period. Am J Obstet Gynecol 153:732-737, 1985.
- Minchom P, Niswander K, Chalmers I. Antecedents and outcome of very early neonatal seizures in infants born at or after term. Br J Obstet Gynaecol 94:431-439, 1987.
- Painter MJ, Scott M, Hirsch RP et al. Fetal heart rate patterns during labor: Neurologic and cognitive development at six to nine years of age. Am J Obstet Gynecol 63:527-532, 1988.
- Holmquist P, Svenningsen NW, Ingemorsson I. Neurodevelopmental outcome and electronic fetal heart rate monitoring in a neonatal intensive care population. Acta Obstet Gynecol Scan 63:527-32, 1984.
- Westgren M, Holmquist P, Ingemorsson I, Svenningsen N. Intrapartum fetal acidosis in preterm infants: Fetal monitoring and long-term morbidity. Obstet Gynecol 63:355-359, 1984.
- Lumley J. Does continuous intrapartum fetal monitoring predict long-term neurological disorders? Paed Perinat Epidemiol 2:299-307, 1988.
- Chalmers I. Randomized controlled trials of fetal monitoring 1973-77. In: Thalhammer O, Bannigarten K, Pollak A (eds). Perinatal Medicine. Stuttgart: Georg Thieme, pp. 260-265, 1979.
- MacDonald D, Grant A, Sheridan-Pereira M et al. The Dublin randomized control trial of intrapartum fetal heart rate monitoring. Am J Obstet Gynecol 152:524-539, 1985.
- Grant A, O'Brien N, Joy M et al. Cerebral palsy during children born during the Dublin trial of intrapartum monitoring. Lancet 2:1233-1236, 1989.
- Grant A. Epidemiological principles for the evaluation of monitoring programs—the Dublin experience. Clin Invest Med 16:149-158, 1993.
- Giles WB, Trudinger BJ, Baird PJ. Fetal umbilical artery flow velocity waveforms and placental resistance: Pathological correlation. Br J Obstet Gynaecol 92:31-38, 1985.
- Trudinger BJ, Stevens B, Connelly AN. Umbilical artery flow velocity waveforms and placental resistance. The effect of embolization of the umbilical circulation. Am J Obstet Gynecol 157:1443-1448, 1987.
Perinatal Asphyxia and Placental Pathology
Carolyn M. Salafia, M.D.
Perinatal Pathology Section, Perinatology
Research Branch, National Institute of
Child Health and Human Development,
Bethesda, Maryland
Introduction
Studies of human infants have identified maternal, fetal, and neonatal factors related to an increased risk of long-term neurologic compromise.1,2 Animal models further elucidate the pathophysiology causal to antenatal brain damage.3,4 Other papers in this volume have summarized present knowledge regarding the scientific basis of brain injury in acute perinatal asphyxia. The Collaborative Perinatal Study has identified a plethora of risk factors for poor long-term neurologic outcome. As summarized by Naeye,2 they are:
- Familial factors (including genetic and environmental);
- Fetal factors (including decreased fetal movement, intrauterine growth retardation, multiple gestations, and fetal anomalies);
- Intrapartum factors (including birth asphyxia, prematurity, passage of meconium, and respiratory distress syndrome);
- No intrapartum or postnatal explanations for brain dysfunction in the presence of antenatal brain maldevelopment or damage.
Risk factors for antenatal brain maldevelopment constitute a diverse group, including several maternal medical diseases, maternal anemia, reduced weight gain in pregnancy, maternal cigarette smoking, and a variety of placental and umbilical cord abnormalities, from single umbilical artery to accessory placental lobes. Whatever the type of neurologic damage seen in the child, one or more risk factors were identified in from 70 to 93 percent of the damaged children.2 However, Kitchen and colleagues have shown identical rates of risk factors in children with and without cerebral palsy" Currently identified risk factors are too poorly specific to permit effective intervention. Also, risk factors may not mark risk for future damage, but indicated preexisting fetal damage. For example, decreased fetal movement in the mid trimester is a risk factor for cerebral damage, but it may mean that cerebral damage has already occurred. Intervention at this point may be too late. We, therefore, need to improve the predictive value of placental factors either necessary for or sufficient to produce poor outcome.
To oversimplify, basic research has identified two major pathways causal to perinatal and neonatal cerebral damage:
- Metabolic insults such as systemic acidosis;4 and
- Hemodynamic insults such as hypoperfusion/reperfusion events.6
The definition of perinatal asphyxia is problematic and is the goal of this workshop. There is clear evidence of the contribution of hypoxia and acidosis to intraventricular hemorrhage.7 However, intrapartum asphyxia, at least as defined by metabolic acidosis, is infrequently identified in the neurologically compromised child.4 Transient changes in fetoplacental hemodynamics may be difficult to document.
Studies of antepartum fetal heart rate monitoring have indicated:
- Poor predictability for major antepartum, intrapartum or postpartum complications;8
- More severely damaged children appearing near[ly] normal in the intrapartum and immediate post-partum period;9 and
- No fall in prevalence rate of cerebral palsy during our present era of fetal heart rate monitoring.
Since evidence of intrapartum insults in brain damaged children is uncommon, the following alternative causes of neurologic damage in the fetus and early newborn period should be sought:
- New types of acute insults, identified when we more completely appreciate and more rigorously monitor the complex fetal and placental transition from intrauterine to extrauterine life;
- Chronic subclinical insults, antedating birth by weeks to months and occurring without clinically recognized maternal or fetal signs or symptoms; and
- Chronic plus acute injury, in which case a chronically suboptimal intrauterine environment sets up a fetus for acute decompensation during the stress of a clinically unremarkable birth process.
We can consider these possibilities in light of placental anatomy and function, and generate for each a set of placental lesions which could contribute to acute compromise, clinically silent antenatal insults, or chronic intrauterine deprivation with acute decompensation.
Pertinent Placental Anatomy and Physiology
The reader is referred to An Introduction to Fetal Physiology for elaboration of the data and hypotheses presented in this review of intrauterine metabolism and nutrient/gas exchange. Battaglia and Meschia11 have summarized the factors upon which net fetal transfer of nutrients depends: "without attempting to make this subject unnecessarily complex, … properties of the placenta, such as maternal and fetal blood flows, the pattern of placental perfusion, the surface, thickness and physicochemical properties of the placental membrane, the metabolic activity of the placenta, the various mechanisms of transfer which are available (e.g., diffusion, carrier mediated transfer, active transfer), and regional differences in placental histology and function."
In the most basic physiologic terms, placental function as a nutrient exchanger requires three components: maternal blood flow, fetal- placental blood flow, and placental trophoblast membrane permeability. Any changes reducing maternal or fetal blood flow, or changing surface area or thickness of the vasculosyncytial membrane, will affect placental exchange functions.
Maternal Blood Flow
Maternal utero-placental flow involves an extensive remodeling of the maternal vasculature, in a process which is complete by the midtrimester.12 Placental trophoblasts normally erode the muscular and elastic tissues of the spiral arteriole to the depth of the inner one-third of the myometrium. The resultant maternal vessel has an enormously increased caliber, and is not responsive to vasomotor stimuli. These vessels permit the normally massive increase in blood flow to the pregnant uterus to occur without increased vascular resistance. The maternal vasculature terminates in the basal plate. Maternal blood is emptied into a trophoblast-lined pool, the intervillous space. Due in part to the flow pattern of maternal blood, placental transfer of nutrients into fetal blood appears to be proportional to the concentrations of nutrients in the maternal blood. The human placenta is an example of a relatively inefficient type of exchanger, a venous equilibrator, and is contrasted to more efficient structures, such as the countercurrent exchangers in the loop of Henle.
Pregnancy compromise has been associated with abnormal conversion of the spiral vasculature in the early months of gestation. In preeclampsia, as well as in idiopathic intrauterine growth retardation in non-hypertensive mothers, abnormal, incomplete or failed trophoblast conversion of the vascular bed can be observed in placental bed biopsies, as well as in decidual vessel segments delivered with the placenta. The persistence of muscular and elastic elements within the vessels results in abnormal uterine vascular resistance and capacitance. This may result in decreased total uterine vascular flow (and therefore nutrient and oxygen presentation) to the placenta. Flow may also be at a greater pressure, and be more turbulent. Reduced flow or flow at an abnormal pressure may affect the developing placental capillary bed. It is important to recall that fetal effects of preeclampsia (e.g., IUGR) may not manifest for weeks to months after uterine vascular conversion is essentially complete (approximately 22 to 24 weeks).
Two additional categories of pathology may compromise the maternal vasculature. Maternal hypercoagulable states are associated with increased risk of pregnancy wastage, based on intrauterine thrombosis and placental infarction. Serologic definition of these states is controversial,13,14 but include hypercoagulability in the context of maternal auto-immune diseases such as systemic lupus erythematosus, and hypercoagulability in otherwise clinically healthy patients. The lupus anticoagulant which was initially identified in patients with systemic lupus erythematosus, may be present and also anti-cardiolipin or anti-phospholipid antibodies in both lupus patients as well as in clinically healthy individuals. The term lupus anticoagulant is a misnomer, as it and its related molecules predispose to thrombosis. The mode of action of these antibodies in the generation of thrombi is also controversial. These antibodies may form immune complexes and initiate coagulation, disrupt prostanoid biosynthesis and/or activate proteins C and S or thrombin, or may be epiphenomena reflecting the presence of a second actually deleterious antibody.15 The uterine vasculature often appears to be particularly susceptible to the thrombogenic properties of these molecules. The uterine vasculature is, of course, unique in that its endothelium is normally eroded, its basement membrane and decidual stromal collagen normally exposed to circulating maternal platelets, and this remodeling takes 20 to 24 weeks to approach completion. It may not be surprising that fetal death associated with antiphospholipid antibodies typically involves decidual thromboses and placental infarctions, and typically occurs before the third trimester.
Toxic and environmental factors may also affect maternal vasomotor tone. Two examples are maternal smoking and maternal cocaine use. Maternal smoking and increased circulating levels of nicotine result in uterine vasoconstriction and reduced uteroplacental flow which may persist for several minutes after cessation of smoking.16 Therefore, while maternal smoking compromises the quality of maternal blood by increasing the concentration of thiocyanate, carboxyhemoglobin and potential carcinogens, it also intermittently and repetitively reduces the quantity of maternal blood presented to the placenta. Cocaine is an even more powerful vasoconstrictive agent. There is an increased incidence of abruption,17 a decidual vascular accident, in mothers using cocaine. Placental transfer of cocaine may also disturb fetoplacental hemodynamics. Cocaine use may be associated with cerebral damage or dysgenesis.18
Placental Hemodynamics
The placental macrovasculature of umbilical and chorionic vessels develops early in gestation. The placental microvasculature enlarges dramatically throughout gestation with the progressive arborization of the villous tree. The factors which regulate placental growth and development of the placental capillary bed are poorly understood. We have observed insulin-like growth factor I and insulin-like growth factor binding protein III to be significantly related to placental weight.19 The placental vasculature increases in complexity until about 36 weeks gestation.20 Factors regulating angiogenesis and vasculogenesis are poorly understood. It is believed that, early in pregnancy, villous endothelium is derived from differentiating mesenchymal cells. Late in pregnancy vasculogenesis appears to occur by proliferation of existing cells rather than by recruitment of differentiating cells. There appears to be a specific villous proliferative zone which may largely determine placental growth at term. Additionally, it is important to recognize that about 40 percent of the placental mesenchymal cells share one or more macrophage markers.21 Since macrophages may induce vasculogenesis, it has been proposed that placental macrophages may contribute to remodeling of the villous capillary bed.22 Abnormal macrophage activity (such as in chronic villitis) may promote abnormal vascular development. The decidua also contains immunocompetent maternal macrophages which could contibute as well to regulation of the growth of the placental vasculature.
Development of the placental vasculature may be abnormal when fetoplacental genetic material is abnormal (i.e., aneuploidy),23 but more commonly is related to maternal states which suggest either restriction of growth by nutrient deprivation (e.g., preeclampsia) or overstimulation of growth (e.g., maternal diabetes mellitus) Scarred, shrunken and fibrotic villi, with reduced number and/or calier of placental capillaries, are commonly observed in association with uterine vascular insufficiency. This may represent destruction of normally forming placental vasculature of impaired development of placental vasculature. Capillary damage may be caused by abnormal force or turbulence of maternal intervillous flow. Midtrimester fetomaternal hemorrhages occur more fequently in hypertensive pregnancies even though placental capillaires are buried deep within the villus.24 Placental capillary breakage may be more frequent in hypertensive pregnancies. Alternatively, chronic uterine vascular insufficiency may lead to decreased placental vascularity by restricting nutrient transfer and reducing the production of growth promoting stimuli. The shrunken, scarred villi may be thought of as the placental equivalent of emphysema. Reduced numbers of vasculosyncytial membranes, and reduced placental exchange efficiency. A reduction in the number and size of villous capillaries would increase placental resistance and fetal cardiac work, since 500ml/min of maternal cardiac output is directed to the placenta. An indirect refelection of placental resistance is the umbilical systolic/diastolic ratio. The systolic/diastolic ratio may approach infinity and negative enddiastolic flow may be seen in cases in which the placental capillary bed is extremely reduced in size.25 In the setting of decreased fetoplacental volume, oligohydramnios may also be present due to reduced glomerular flow; oligohydramnios is a marker for risk of fetal distress.26
Chorangiosis, and excessive proliferation of small caliber, tortuous vessels, is seen in association with maternal diabetes mellitus, a condition in which growth factor production and regulation may be altered. Increased capillary proliferation is observed in maternal smoking and has been linked to unexplained intrauterine growth retardation. The underlying causes, however are unclear. A purely speculative interpretation may be that capillary proliferation represents a dysfunctional attempt by the placenta to compensate for inadequate fetal or placental nutrition. This may explain why capillary proliferation is common in twin gestations, in which two fetuses are competing for a single volume of uterine blood flow. When there is excessive capillary proliferation, placental nutrient exchange functions may be affected because many of the vessels are deep within the villus, and there may not be efficient nutrient exchange areas. Placental microvascular resistance, fetoplacental intravascular volume, and therefore fetal cardiac work may all be increased. However, even in cases with extensive capillary proliferation and tortuousity, umbilical systolic/diastolic ratios may remain within normal limits. Chorangioma is a focal placental vascular tumor or malformation, which may occur singly or multifocally and functions as an arteriovenous malformation or vascular shunt. This lesion may present with stillbirth, hydrops fetalis due to congestive heart failure, or unexplained fetal tachycardia.
Villous Surface Exchange Area
The villous surface area of exchange increases throughout gestation, to compensate for increasing fetal metabolic demands. Placental permeability increases even after the maximal rate of placental growth plateaus. In the human, this observation can be explained by increased numbers of vasculosyncytial membranes. Earlier in pregnancy the placental capillary is deep within the villus; by term many villous capillaries abut the trophoblast basement membrane, with trophoblast nuclei clustering at the side. The vasculosyncycial membrane measures only 0.5 to 1.0 micrometer in thickness. Within it, the capillary basal lamina and trophoblastic basal lamina may actually fuse,28 leaving only minimal impediments to nutrient diffusion and gas exchange. Additionally the microvillous surface increases until about 36 weeks gestation with microvillous surface area actually being reduced in the last 4 weeks of pregnancy.29 The progressive development of optimal placental permeability with gestational age leads to a potentially flow-limited placental transfer capacity at term.
Oxygen and carbon dioxide are exchanged across the placenta by diffusion. However, the placenta is a very metabolically active tissue, consuming almost 50 percent of the O2 presented to the intrauterine tissues. From fetal oxygen utilization one can calculate the fetal metabolic rate. Fetal metabolism is directed to build new tissues and energy stores and to support energy demands of existing tissues. The fetus may direct placentally supplied nutrients, including oxygen, to different tissue uses at different gestational ages. Fetal CO2 production is approximately equal to PO2 utilization. Fetal CO2 diffuses across the placenta rapidly into the maternal blood based on the concentration gradient between fetal and maternal blood. Fetal PCO2 may drop with maternal hyperventilation, and rise with maternal hypoventilation, impaired placental perfusion, or impaired placental permeability. Fetal arterial PCO2 can rise due to fetal under-perfusion of the placenta (or cord compression).
The fetus has no stores of oxygen; all oxygen is supplied by maternal blood. For most other nutrients, including glucose, there is fetal or fetoplacental reserve. The placenta metabolizes much of the glucose obtained from the maternal circulation. The amount of molecular glucose provided to the fetus by the placenta is less than the amount of substrate required to fuel the fetus. Glucose transfer across the placenta is a carrier-mediated event, affected by changes in perfusion. A major placental metabolite of glucose is lactate. Most lactate throughout gestation is provided by the placenta and is a major fetal substrate, since supplied glucose must be reserved for the developing central nervous system. In intrauterine growth retardation, fetal nutrient utilization may decrease,30 leading to underutilization of placental lactate and a lactic acidosis. Under conditions of asphyxia the fetus itself is a net producer of lactate. Ketoacids may reduce fetal brain metabolism.
In summary, the placenta functions as an imperfect venous equilibrator, transferring molecules into the fetoplacental pool proportionate to their concentrations in the maternal circulation. Features which limit fetal oxygen and nutrient supply, and therefore fetal metabolism, are: Changes in maternal perfusion;
- Changes in fetoplacental blood flow;
- Changes in placental permeability; and
- Placental metabolic demands.
The principal maternal adaptation favoring fetal oxygenation is a high uterine blood flow. Uniform fetal perfusion of the placenta, production of a fetal hemoglobin with greater oxygen affinity than maternal hemoglobin, and a high fetal cardiac activity and increased fetal tissue perfusion are the main mechanisms by which the fetus optimizes its oxygenation. Normal placental growth and development will maximize placental efficiency while controlling placental metabolic demands.
Placental Anatomy/Physiology and Cerebral Damage
Acute Mechanisms of Brain Damage in the Term Infant
Abnormalities in placental blood flow causing acute fetal brain damage may focus on three different anatomic sites: the umbilical arteries and vein, the chorionic vessels, and the villous capillary bed. The umbilical arteries and vein are the largest volume carriers. With the two arteries wrapped around the vein, low-pressure venous blood is milked back through the cord to the fetus courtesy of arterial pulsations. Cases of single umbilical artery are associated with increased likelihood of intrauterine growth retardation,31 possibly related to subtle compromise of normal cord flow.
Cord compression may cause fluctuations in fetal blood flow. Arterial compression increases placental resistance, fetal cardiac work and fetal myocardial oxygen requirements; thus, umbilical venous compression may produce both hypoxemia (reduced O2 availability) and ischemia (decreased blood flow). In twin gestations intrauterine growth retardation is frequently seen in the co-twin with abnormal cord insertion.32 A greater ease/likelihood of vascular compression may be associated with membranous vessels and velamentous cord insertion. It is likely that the same mechanics could compromise the singleton fetus with a velamentous cord insertion as well. Structural defects have also been appreciated.33 From pure mechanics, it is not difficult to understand how a velamentous cord insertion could compromise fetal placental blood flow. Placental thickness in utero is approximately twice that measured after delivery in the pathology laboratory; active maternal intervillous circulation expands the intervillous space. The chorionic vessels on the chorionic plate are located on a deformable bed of villi and maternal blood. The vessels which run in the extraplacental membranes are apposed directly to the rigid myometrial wall. Any compression of these vessels will reduce flow, damage their musculature and/or endothelium and lead to thrombosis, thromboemboli or chronically abnormal flow. Thromboemboli originating in the placental arterial tree may lodge in the placental capillary bed, occluding the circulation and compromising placental function. More significant to the fetus are venous-derived placental thromboemboli which may spread into the fetal cerebral circulation, occluding vessels with irreparable consequences.
Trauma and other mechanical events are not the only stimuli for generation of thrombi. Inflammation activates endothelium to promote platelet aggregation. Both acutely and chronically inflamed vessels of the placental tree may demonstrate mural thrombi, and may predispose to thromboembolic events. Inflammatory processes may also involve production of locally generated and systemic vasomotor stimuli, generally arachidonic acid derivatives. These substances can induce vasoconstriction or vasodilatation at various levels within the placental vasculature.34 Acute ascending, often bacterial, invasion of the amniotic fluid space leads to intraamniotic accumulation of cytokines and other inflammation-related molecules which diffuse into the placental tissues and vascular structures with resultant vasomotor effects. Also, structural changes in the umbilical vessels accompany inflammation. The vascular muscle may become edematous and the endothelium may balloon; these changes may indicate altered vessel function. Increased umbilical systolic/diastolic ratios have been observed in human fetuses with inflamed umbilical vessels in their delivered placentas.36 These vasoactive molecules also regulate blood flow in smaller branches of the placental vasculature.
Within the microcirculation of the villous capillary bed, tissue hypoxia has been shown to have a potent vasoconstrictive effect on regional placental vasculature,37 effectively shunting blood away from placental regions poorly perfused by maternal blood, and directing blood to areas of the placenta better supplied with oxygen and nutrients. This is analogous to the ventilation/perfusion mismatch which may lead to respiratory insufficiency. Therefore, at all levels of the placental circulation, resistance, capacitance and flow may be acutely modulated by mediators generated in intraamniotic infection and by variations in maternal perfusion leading to regional placental hypoxia.
It is known that in an abruption, a decidual vascular accident causes retroplacental hemorrhage which traumatically detaches the placenta from the uterine lining. This acute event may be due to acute vascular damage related to cocaine use. Wigglesworth has eloquently described the hemodynamic and hypoxic effects of abruption on the fetus. An area of placental tissue is acutely rendered incapable of participating in nutrient exchange, leading to acute changes in fetal oxygen availability while placental compression increases fetal placental blood pressure, predisposing to visceral hemorrhage.6 However, function of areas distant from the abruption may also suffer due to changes in perfusion and resistance of the fetoplacental unit as a whole.
In our current approach, we may be underestimating the extent of altered placental function, and overlooking more general placental dysfunction. A histologic marker for placental dysfunction may be villous edema. Hemodynamic shifts may acutely affect nutrient exchange and fetal perfusion. Villous lesions, which theoretically may affect nutrient exchange, increase the diffusion distance for nutrients across the placenta, and thus impair placental permeability. These include thickening of the trophoblast basement membrane, seen in diabetes mellitus,38 reduced total area of vasculosyncytial membranes as may be seen in preeclampsia and villous edema. Villous edema has been described to occur with greater frequency in cases with acute intrauterine inflammation.39 However, acute villous edema may be seen in abruptio placenta, cases of maternal diabetes mellitus, and in cases with unexplained intrapartum distress. In villous edema, fluid accumulates in the villous stroma. Naeye suggested that increased intravillous pressure may collapse the capillary bed and lead to hypoxia.39 This is often very difficult to appreciate at the microscope; edematous villi often have many vessels of normal caliber and may even be congested. Basically any process that affects trophoblast epithelial permeability and/or transport, capillary vasomotor tone or capillary permeability may lead to fluid leakage and villous edema. Villous edema may be a marker for placental dysfunction related to many different reasons, i.e., endotoxin-induced trophoblast damage, placental macrophage activation and cytokine production, or increased capillary perfusion pressure, and therefore may have different implications for the fetus depending upon its etiology.
A related lesion may be villous stromal hemorrhage. This lesion of intraplacental bleeding may be a precursor to fetomaternal transfusion and it is often observed in the vicinity of a placental abruption. It could be viewed as a placental bruise resulting from the trauma of placental detachment from the uterine lining. Any fetal blood loss, whether into the maternal circulation or into the placenta, may destabilize normal fetoplacental hemodynamics. If an abruption initially increases fetal perfusion and blood volume, subsequent placental bleeding may contribute to fetal hypovolemia and generation of hypoperfusion lesions. Villous stromal hemorrhage may be seen in very edematous villi, suggesting that capillary integrity is impaired.
Cerebral Insults Prior to Labor and Delivery
Cerebral insults may occur prior to labor and delivery without clinically recognized maternal or fetal signs and symptoms. Insults antedating birth may include:
- Acute and temporally limited events; and/or
- Chronic disease processes which progressively compromise the fetus with a climax in acute tissue damage prior to birth.
Acute, temporally limited processes would resolve with time, resulting in the birth of an infant with repaired visceral function but persistent irreparable brain damage. Many of the same events which may cause acute intrapartum damage may, if severe enough, cause antepartum cerebral damage. Examples of time-restricted damage processes include:
- Maternal abdominal trauma from any cause leading to abnormal maternal perfusion, maternal hypotension, hypovolemia or shock;
- Placental abruption with delayed delivery and hypoxic and hemodynamic effects involving the viscera but not the central nervous system;
- Umbilical cord compression or vascular compression in membranous cord insertion (see section on Acute Mechanisms of Brain Damage in the Term Infant);
- Spontaneous fetomaternal or fetofetal transfusion, with fetal hypovolemia and hypoperfusion following fetomaternal transfusion or death of a co-sibling; and
- Acute infectious/inflammatory processes which may spontaneously resolve.
Acute time-limited and chronic events may overlap since there is imprecision in histological timing of anteparcum events in the delivered placenta, and also multiplicity of effects of an event on uteroplacental function. For example, in the ewe, uteroplacental embolization may acutely damage the placenta but also alters placental endocrine function (cortisol and progesterone biosynthesis). In this situation, an acute event may result in a more chronic dysfunction.40
There may also be different mechanisms which result in any one placental lesion, so that one lesion could reflect either acute or chronic pathophysiology. Villous stromal hemorrhage has been described as acute intraplacental bleeding, but may also accompany other lesions which have been considered to reflect more chronic placental circulatory instability, including the so-called hemorrhagic endovasculitis accompanied by endothelial damage and intraplacental erythrocyte fragmentation. The triad of villous stromal hemorrhage, intraplacental thrombi and hemorrhagic endovasculitis is not uncommon in cases of unexplained stillbirth, and in cases with unexpected fetal decompensation during the delivery process. Recognition of hemosiderin pigments indicates that circulatory damage and intraplacental bleeding predates birth by a minimum of 24 to 48 hours.41 Likewise a circumvallate insertion of the placental membranes may reflect an acute event (decrease in amniotic fluid pressure) or a chronic process (abnormal depth of placentation).42
Some acute events are the climax of subclinical chronic pathology. The uterine vascular anatomy which suffers a spontaneous acute abruption may have developed abnormally since the early weeks of pregnancy. Women may be normotensive, and their uterine and placental anatomy may show changes of chronic uterine vascular insufficiency similar to those seen in preeclampsia. Maternal hypercoagulable states predispose to decidual thromboses and placental infarction, and can chronically impair the well-being of the fetoplacental unit. However, acute decidual vessel occlusion may lead to vessel rupture and abruption. Cord compression events also may be acute but repetitive. Animal studies suggest that it is the repetitive cord compression which is most effective in the generation of cerebral damage.43
Finally, new pathophysiologic pathways have complicated the timing issue. For instance, the mechanisms of release of nucleated erythrocytes by the bone marrow have multiplied. The traditional pathway of hypoxia inducing erythropoeitin to affect the bone marrow has been invoked to explain the association of excess nucleated erythrocytes in the fetoplacental circulation with increased risk of cerebral compromise.44 However, infection induces both maternal and fetal production of interleukin-6, which has recently been shown to have a direct effect on the bone marrow to stimulate proliferation of immature cells of multiple lineages, including immature (nucleated) erythrocytes.45 In acute intraamniotic infection there are increased fetal circulating levels of interleukin-6. Acute amniotic infection is also a risk factor for cerebral damage.46 Therefore nucleated erythrocytes may be a marker of relatively chronic hypoxia, or may be a manifestation of a fetal systemic response to acute intraamniotic infection. Further studies are needed to untangle the mechanisms underlying the release of nucleated red blood cells from bone marrow in cases with cerebral damage.
In monozygous twins, there are essentially obligatory vascular anastomoses which provide an anatomy in which acute and chronic insults may both be present. Chronic twin transfusion may lead to chronic hypoxia and relative hypovolemia in the donor, and volume overload and congestive heart failure in the recipient. Doppler studies indicate that these hemodynamics may be very dynamic, occasionally observed with reversals of flow.47 Chronic hypoperfusion/reperfusion injury in both twins may be frequent. Acutely, death of a co-twin may result in variable degrees of exsanguination of the living sibling into the dead baby through the circulatory connections. Acute hypovolemic shock may lead to death of the sibling but homeostasis may also occur with resultant visceral but not cerebral repair. In studies of children in whom antenatal cerebral damage is thought to have occurred, many of them are monochorionic twins.48
Certain processes are likely to have primarily chronic effects. Maternal smoking is an example of a cumulative chronic process. We have observed increased capillary proliferation in many placentas of smoking mothers, and others have described lesions which reasonably should affect placental nutrient exchange efficiency, such as basal lamina thickening.49 Additionally, toxic compounds which circulate in the blood of mothers who smoke may accumulate in the fetus and placenta, with cumulative tissue damage effects.50 There are also direct effects of maternal smoking on circulation in the umbilical vessels,51 fetal coronary arteries and aorta.52,53
Maternal diabetes mellitus may also create a situation of chronic ongoing fetal compromise. In placentas delivered of diabetic mothers, there is often diffuse abnormal maturation of the placental villi. Villi are too large, too poorly vascularized, with centrally located vessels (rather than vasculosyncytial membranes), and with immature appearing bilayered trophoblast and prominent stroma. Vasculosyncytial membranes, if present, may be thickened. Diabetic placentas are often larger than normal, and fetoplacental weight ratios slightly decreased, indicating that placental growth may outstrip fetal growth. These disproportionately large organs have disproportionate metabolic demands. Because of this abnormal anatomy these placentas may be more inefficient in nutrient transport. The progressive switch of fetal production of hemoglobin F to hemoglobin A may, especially in the presence of high-oxygen affinity hemoglobin A1C in the maternal circulation, reduce fetal capacity to extract oxygen from the maternal circulation and progressively impair fetal oxygen supply.
I have already discussed maternal hypercoagulable states in the context of decidual vascular pathology. Decidual thromboses and placental infarction can chronically impair the well-being of the fetoplacental unit. We have recently documented increased coagulation on the trophoblast surface (perivillous coagulation) and intraplacental thrombi in cases of antiphospholipid antibody-associated pregnancy compromise.54 These lesions develop despite full anticoagulative therapy. Current therapy, based on attempts to anticoagulate the mother, may only incompletely remedy antiphospholipid antibody-related placental damage. It is reasonable to assume that perivillous coagulation may lead to placental asphyxia, by essentially walling off the placental nutrient exchange surface from maternal flow. An extreme form of perivillous coagulation is called maternal floor infarction. This term is a misnomer, since the histology is not compatible with a necrotic lesion. Villi devoid of trophoblast float in a sea of fibrin admixed with proliferating X-cells (cytotrophoblasts). These lesions have been reported to be recurrent.55 We have much to learn about the means by which the trophoblast epithelium lining the intervillous vascular space may or may not assume all the anticoagulative functions of endothelium. Thrombomodulin may be expressed by the trophoblast (L Zacharski, personal communication). Perivillous coagulation may result from:
- Traumatic, ischemic or other trophoblast damage with exposure of basement Membrane and collagen to circulating maternal platelets;
- Immune complex-mediated trophoblast damage with coagulation similar to the mechanism;
- Direct immune complex-mediated activation of coagulation; and/or
- An intrinsic trophoblast abnormality.
The exact mechanism(s) is unclear. However, when intraplacental thrombi occur due to these chronic processes, they may lead to placental-fetal thromboemboli and acute cerebral damage.
Finally, an all-too-common finding in cases of unexplained fetal/neonatal damage is that of chronic villitis. Chronic villitis, chronic inflammation of the placental villi, is believed to indicate a congenital viral infection.56 Recently chronic villitis has been associated with autoimmune responses of the mother, possibly to the extensive modifications of her tissues by the conceptus, and also to alloimmune rejection of the conceptus. Most cases of congenital viral infection are clinically asymptomatic in the mother. Autoimmunity may also be subclinical, with no evidence of maternal systemic disease, but only serologic abnormalities. Maternal/fetoplacental alloimmune pathology may also be without any recognized maternal syndrome or specific laboratory abnormality. The actual amount of villi inflamed is often quite small, and insufficient to explain fetal compromise on the basis of anatomic placental insufficiency. It is proposed that viral infections are associated with reduced fetal mitosis and decreased cell number. Other effects of chronic inflammation may be changes in placental macrophage activity. Macrophage products may locally affect placental microcirculatory growth, hemodynamics and trophoblast function. Also maternal invasion of the conceptus in villitis may lead to intrauterine graft versus host disease and widespread fetal damage.
Chronic Suboptimal Intrauterine Environment
A chronically suboptimal intrauterine environment may set up a fetus for acute decompensation during the birth process from seemingly mild stress. Intrauterine growth retardation of the fetus is associated with increased risk of cerebral damage. Chronic nutrient deprivation, and a reduced placental exchange area are likely to be present in almost all cases in which there is reduced placental weight. Studies of human birthweight data have indicated that as much as 39 percent of variation in human birthweight can be attributed to genetic or environmental maternal factors, 15 percent to environmental factors unique to each mother, and 46 percent to environmental factors which are unique to each pregnancy.57 It is known in certain animals that paternal genetic information is preferentially expressed in the placenta. Potentially paternal genes may limit placental and secondarily fetal growth. Such cases of intrauterine fetal and placental growth retardation may not be amenable to therapy. Growth-impaired infants as a group are more susceptible to developing clinical distress during the birth process, as an acute decompensation.
Alternatively, a chronically abnormal intrauterine environment may compromise a genetically competent fetoplacental unit. I have discussed a number of chronic and often subclinical maternal and placental diseases which may result in antepartum tissue damage or may be associated with adequate compensatory adaptations by the fetus (such as decreased fetal growth) without the development of actual antepartum tissue damage. The antepartum compensation of the fetus may, however, limit its adaptability to the stress of a clinically normal labor and delivery. Acute mechanical effects on umbilical cord or velamentous vessels, acute infectious stress, or simply the stress of repetitive placental breath-holding during uterine contractions, may become the last straws on the camel's back of chronic fetal compromise, and present as an unexplained acute decompensation.
Placental Histology and Timing of Antepartum Insults
Significant improvements in our ability to time the onset of clinically silent histological disease processes in the placenta would be a major boon. In contrast to the opinion expressed by American College of Obstetricians and Gynecologists (Placental Pathology, June 1993), most placental lesions and disease processes are clinically silent! For example, a woman presents with a 4-hour history of back pain, fails tocolysis, rapidly progresses and delivers 4 hours later. The histology indicates extensive polymorphonuclear leukocyte infiltration with many fragmented polymorphonuclear leukocytes. The lifespan of the adult polymorphonuclear leukocyte is approximately 1 to 2 days after diapedesis. The histology suggests that an intraamniotic inflammation event was initiated prior to the onset of any clinical symptoms.
Meconium staining has been studied in one in vitro experiment. This study suggested that meconium phagocytosis by amniotic macrophages occurred within 1 to 3 hours, and meconium phagocytosis by chorionic macrophages occurred within 3 to 6 hours. Staining of the umbilical cord and decidua has not been studied.
I have discussed nucleated erythrocytes in the context of cerebral damage. In the adult, generally a hypoxic stress requires 24 to 48 hours for detection of circulating nucleated erythrocytes. This time course must vary in the fetus due to the following:
- Gestational age, with younger ages having greater extramedullary hematopoeisis and facilitated egress of nucleated erythrocytes into the circulation;
- Duration and severity of stress, with protracted duration promoting excessive extramedullary hematopoeisis, and a more severe stress directly damaging the bone marrow; and
- Pressures of additional factors affecting bone marrow gating of erythrocytes such as interleukin-6 in acute infection).
We have mentioned that metabolism of hemoglobin to hemosiderin is considered to take a minimum of 24 to 48 hours. This has been useful in dating intrauterine bleeding processes, from chronic to acute abruption, to hemorrhagic endovasculitis/circulatorydamage states, to umbilical cord hemorrhages, but cannot extend the timing beyond 1 to 2 days before delivery of the placenta.
Trophoblast degenerative knots may develop as quickly as 24 hours after in vitro exposure to lowered oxygen tension. However, trophoblast knots are also common in situations of chronic uterine vascular insufficiency, such as preeclampsia. Trophoblast knots which develop as a response to an acute stimulus are associated with normal appearing villi. Trophoblast knots appearing as part of a more chronic process will have abnormal villous structure, including fibrosis and hypovascularity. Infarctions with loss of nuclear detail will likely be more than 7 days old.
The age of chronic villitis may be a more complicated issue. The premature placenta, with its functionally immature macrophages, may be less capable of demonstrating a cellular infiltrate than a term placenta. Small foci of villitis in a precerm placenta may represent a significantly more severe and protracted inflammation than in a term placenta. The possibility that inflammation may present in a different fashion in the very immature placenta should also be considered. Perhaps in the very immature placenta, a cellular infiltrate will not develop. Instead, edema, a cardinal sign of inflammation, may be the only histologic evidence of villous inflammation. The gestational age of the placenta at the time of insult and at delivery may affect placental tissue response to intrauterine events, and placental-fetal pathophysiology.
Summary
The search in the intrapartum period for the roots of every case of neurological damage has not been rewarding. Processes related to long-term compromise of neurologic function are usually clinically silent. Given what is understood of placental function, and pathways which could lead to cerebral damage, I have proposed specific anatomic sites in which disturbance of anatomy or changes in functional capacity could contribute to the initiation of metabolic and hemodynamic processes which lead to cerebral damage.
Light microscopic studies are not likely to yield diagnoses which alone may be sufficient and specific enough to explain cerebral damage. Lesions identified by light microscopy may point to clinically silent pathways of fetal damage, or may lend support to relevant clinical and biochemical clues. Computer-assisted image analyses (to better quantify and standardize diagnoses of lesions) and immunohistochemistry and in situ hybridization (to more completely describe and better characterize lesions) may become necessary to supplement our present diagnostic capabilities.
References
- Nelson KB, Ellenberg JH. The asymptomatic newborn and risk of cerebral palsy. Am J Dis Child 141:1333, 1987.
- Naeye RL. Disorders of the placenta, fetus, and neonate: Diagnosis and clinical significance. St. Louis: Mosby Year Book, Inc., 1992.
- Low JA, Galbraith JS, Muir DW et al. Factors associated with motor and cognitive deficits in children after intrapartum fetal hypoxia. Am J Obstet Gynecol 148:533, 1984.
- Low JA, Froese AF, Galbraith RS et al. The association of fetal and newborn metabolic acidosis with severe periventricular leukomalacia in the preterm newborn. Am J Obstet Gynecol 162:977-982, 1990.
- Kitchen WH, Doyle LW, Gord GW et al. Cerebral palsy in very low birth weight infants surviving to two years with model prenatal intensive care. Am J Perinatol 4:29-35, 1987.
- Wigglesworth JS, Pape K. Haemorrhage, ischemia and the perinatal brain. Early Hum Dev 2:179-188, 1978.
- Leech RW, Kohnen P. Subependymal and intraventricular hemorrhages. Am J Pathol 77:465-476, 1974.
- Goldkrand JW, Benjamin DS. Antepartum fetal heart testing: A clinical appraisal. Obstet Gynecol 63:48-51, 1984.
- Hull J, Dodd KL. Falling incidence of hypoxic-ischemic encephalopathy in term infants. Br J Obstet Gynaecol 99:386-391, 1992.
- Lamb B, Lang R. Aetiology of cerebral palsy. Br J Obstet Gynaecol 99:176-178, 1992.
- Battaglia FC, Meschia G (eds). An Introduction to Fetal Physiology. London: Academic Press Inc., 1986.
- Pijnenborg R, Dixon G, Robertson WB, Brosens I. Trophoblastic invasion of human decidua from 8-18 weeks of pregnancy . Placenta 1:3- 19, 1980.
- Feinstein DI, Lupus anticoagulant, thrombosis, and fetal loss. N Engl J Med 21:1348, 1985.
- Cowchock S, Smith JB, Gocial B. Antibodies to phospholipids and nuclear antigens in patients with repeated abortions. Am J Obstet Gynecol 155:1002-1010, 1986.
- Triplett DA. Symposia - Pathogenic mechanisms of action: Effects on the coagulation system. Fifth International Symposium on Antiphospholipid Antibodies, San Antonio, Texas, 1992.
- Andersen KV, Hermann N. Placenta flow reduction in pregnant smokers. Acta Obstet Gynaecol Scand 63:707-709, 1984.
- Dombrowski MP, Wolfe HM, Welch RA, Evans MI. Cocaine abuse is associated with abruptio placentae and decreased birth weight, but not shorter labor. Obstet Gynecol 77:139-141, 1991.
- Chasnoff IJ, Bussey ME, Savich R, Stack CM. Clinical and laboratory observations: Perinatal cerebral infarction and maternal cocaine use. J Pediatr 108:456-461, 1986.
- Fant M, Salafia CM, Baxter RC et al. Circulating levels of IGFs and IGF binding proteins in human cord serum: Relationship to intrauterine growth. Regulatory Peptides 48:29-39, 1993.
- Teasdale F. Gestational changes in the functional structure of the human placenta in relation to fetal growth: A morphometric study. Am J Obstet Gynecol 137:560-568, 1980.
- Goldstein J, Braverman M, Salafia CM, Buckley PJ. The phenotype of human placental macrophages and its variation with gestational age. Am J Pathol 133: 648, 1988.
- Demir R, Kaufmann P, Castelucci M et al. Fetal vasculogenesis and angiogenesis in human placental villi. Acta Anat 136:190-203, 1989.
- Rochelson B, Kaplan C, Guzman E et al. A quantitative analysis of placental vasculature in the third trimester fetus with autosomal trisomy. Obstet Gynecol 75:59-63, 1990.
- Los FJ, deWolf BTHM, Huisjes HJ. Raised maternal serum-alpha fetoprotein levels and spontaneous fetomaternal transfusion. Lancet 2:1210-1212, 1979.
- Giles WB, Trudinger BJ, Baird PJ. Fetal umbilical artery flow velocity waveforms and placental resistance: Pathological correlation. Br J Obstet Gynaecol 92:31-38, 1985.
- Groome LJ, Owen J, Neely CL, Hauth JC. Oligohydramnios: Antepartum fetal urine production and intrapartum fetal distress. Am J Obstet Gynecol 165:1077-1080, 1991.
- Altshuler G, Herman AA. The medico-legal imperative: Placental pathology and epidemiology Stevenson DK, Sunshine P (eds). In: Fetal and Neonatal Brain Injury: Mechanisms, Management, and the Risks of Practice. B.C. Decker Inc., pp. 250-263, 1989.
- Benirschke K, Kaufmann P. Pathology of the Human Placenta, 2nd Edition, Springer-Verlag, pp. 29, 1990.
- Teasdale F. Jean-Jacques G. Morphometric evaluation of the microvillous surface enlargement factor in the human placenta from mid- gestation to term. Placenta 6:375-381, 1985.
- Clapp JF. Szeto HH, Larrow R et al. Fetal metabolic response to experimental placental vascular damage. Am J Obstet Gynecol 140:446-451, 1981.
- Benirschke K, Kaufmann P. Pathology of the Human Placenta, 2nd Edition, Springer-Verlag, pp. 209, 1990.
- Official Proceedings of the 37th Annual Convention, American Institute of Ultrasound in Medicine, Volume 12, No. 3 (supplement), March 1993.
- Benirschke K, Kaufmann P. Pathology of the Human Placenta, 2nd Edition, Springer-Verlag, pp. 716, 1990.
- Mak K K-W, Gude NM, Walters WAW, Boura ALA. Effects of vasoactive autocoids on the human-fetal placental vasculature. Br J Obstet Gynaecol 91:99-106, 1984.
- Howard RB, Hosokawa T, Maguire MH. Pressor and depressor actions of prostanoids in the intact human fetoplacental vascular bed. Prostaglandins Leukotrienes Med 21:323-330, 1986.
- Fleming AD, Salafia CM, Vintzileos AM et al. The relationships amongst umbilical arterial velocimetry, fetal biophysical profile and placental inflammation in preterm premature rupture of the membranes. Am J Obstet Gynecol 164:38-41, 1991.
- Howard RB, Hosokawa T, Maguire MH. Hypoxia induced fetoplacental vasoconstriction in perfused human placental cotyledons. Am J Obstet Gynecol 157:1261-1266, 1987.
- Benirschke K, Kaufmann P. Pathology of the Human Placenta, 2nd Edition, Springer-Verlag, pp. 478, 1990.
- Naeye R, Maisels J, Lorenz RP, Botti JJ. The clinical significance of placental villous edema. Pediatr 71:588-594, 1983.
- Clapp JF, Auletta FJ, Farnham J et al. The ovine fetoplacental endocrine response to placental damage. Am J Obstet Gynecol 144:47-54, 1982.
- Kissane JM (ed). Anderson's Pathology, 6th Edition. St. Louis: CV Mosby Co., Volume 1, pp. 88, 1971.
- Benirschke K, Kaufmann P. Pathology of the Human Placenta, 2nd Edition, Springer-Verlag, pp. 386-391, 1990.
- Clapp JF, Peress NS, Wesley M, Mann LI. Brain damage after intermittent partial cord occlusion in the chronically instrumented fetal lamb. Am J Obstet Gynecol 159:504-509, 1988.
- Altshuler G, Herman AA. Placenta within the medicolegal imperative. Arch Pathol Lab Med 115:688-695, 1991.
- Heinrich PC, Castell JV, Andus T. Review article: Interleukin-6 and the acute phase response. Biochem J 265:621-636, 1990.
- Naeye RL. Amniotic fluid infections, neonatal hyperbilirubinemia, and psychomotor impairment. Pediatrics 62:497-503, 1978.
- Pretorius DH, Manchester D, Barkin S et al. Doppler ultrasound of twin transfusion syndrome. J Ultrasound Med 7:117-124, 1988.
- Bejar R, Vigliocco G, Gramajo H et al. Antenatal origin of neurologic damage in newborn infants. Am J Obstet Gynecol 162:1230-1236, 1990.
- Burton GJ, Palmer ME, Dalton EJ. Morphometric differences between the placental vasculature of non-smokers, smokers, and ex-smokers. Br J Obstet Gynaecol 96:907-915, 1989.
- Kuhnert PM, Kuhnert BR, Bottoms SF, Erhard P. Cadmium levels in maternal blood, fetal cord blood, and placental tissues of pregnant women who smoke. Am J Obstet Gynecol 142:1021-1025, 1982.
- Busacca M, Balconi G, Pietra A et al. Maternal smoking and prostacylcin production by cultured endothelial cells from umbilical arteries. Am J Obstet Gynecol 148:1127-1129, 1984.
- Lehtovirta P, Pesonen E, Sarna S. Effect of smoking on the fetal coronary arteries. Acta Path Microbiol Immunol Scan Sect A 92:189-193, 1984.
- Eriksen PS, Marsal K. Circulatory changes in the fetal aorta after maternal smoking. Br J Obstet Gynaecol 94:301-305, 1987.
- Parke AL, Salafia CM, Ernst L et al. Placental pathology in patients with phospholipid antibodies. Abstract, presented at the Fifth International Symposium on Antiphospholipid bodies, 1992.
- Clewell WH, Manchester DK. Recurrent maternal floor infarction: A preventable cause of fetal death. Am J Obstet Gynecol 147:346-347, 1983.
- Altshuler G, Russell P. The human placental villitides: A review of chronic intrauterine infection. In: Current Topics in Pathology. Heidelberg: Springer-Verlag, 1975.
- Nance WE. 1992 American Society of Human Genetics presidential address: Back to the future. Am J Hum Genet 53:6-15, 1993.
Discussion
Nigel Paneth, M.D., M.P.H.
Michigan State University, East Lansing
The three papers just presented raise an interesting but infrequently addressed question about acute perinatal asphyxia, namely, when and where is asphyxia optimally assessed and/or measured? Each of the three authors takes a different segment of the maternal-placental-fetal unit and explores its relationship, actual or potential, to acute perinatal asphyxia.
Evidence of asphyxia can be obtained from several anatomic locations and at several points in time, each with its own limitations and advantages. Sampling of the umbilical artery in utero for blood gases may be a scientifically valid approach to quantitating asphyxia, but is impractical in most circumstances, and impossible during labor. Information from the placenta cannot guide clinical management, because its major pathologic features are observable only after birth. Fetal heart rate tracings are proxy measures of asphyxia filtered through the variability of individual fetal response. Although easy to obtain, their scientific validity as measures of asphyxia is suspect. The optimum compromise between clinical utility and scientific validity is what defining acute perinatal asphyxia is all about.
Dr. Salafia provides an overview of placental anatomy physiology, and biochemistry, emphasizing the complexity of this organ, and the multiplicity of placental pathophysiologies capable of producing either acute or chronic perinatal asphyxia. Unfortunately, our understanding of the relative importance of any of these mechanisms is very limited, since the prevalence of these conditions in the population, and their relationship to measures of acute perinatal asphyxia in the fetus or newborn, are largely unknown.
Placental dysfunctions most clearly lead to acute asphyxia by vascular mechanisms; placental abruption and cord prolapse are two well-known examples. Chorionic vessels and the villous capillary bed can also be sites of obstructive thromboses or inflammatory induced vasoconstriction, leading to acute asphyxia. Dr. Salafia also describes some of the placental findings, such as villous edema or villous stromal hemorrhage, that may indicate a fetus at risk for acute perinatal asphyxia. But the author notes that there is substantial lack of specificity to many pathological indicators of placental damage. Any number of insults, ranging from infection to trauma, may produce similar placental pathology. Thus the precise role of the placenta in helping to define acute perinatal asphyxia remains obscure. And, although we often view the placenta as a source of evidence that chronic asphyxia antedated the onset of labor, we are reminded of the "imprecision in histological timing of antepartum events in the delivered placenta" (Salafia).
The placenta provides a background of information that can assist interpretation of the sequence of events leading to acute perinatal asphyxia. Badly needed are quantitative studies that attempt to correlate the placental findings with indicators of acute perinatal asphyxia in the fetus, to better understand how the placental environment contributes to or mediates the risk of acute perinatal asphyxia.
Dr. Low reviews studies that examine blood flow velocity, determined by Doppler ultrasound techniques on either side of the placenta—in the uterine and umbilical arteries. In general, uterine artery flow velocity seems less closely related to the status of the fetus than does umbilical artery flow velocity, although fewer studies address this maternal vascular measure. A modest correlation of umbilical artery flow velocity with fetal distress has been found in most studies, but the positive predictive value of an abnormal systolic/diastolic ratio is low, and there is substantial overprediction of distress. With the exception of one study of nine fetuses, correlations with antepartum blood gas values are also modest.
A yet more mixed picture emerges when the outcome of interest is cord blood gases at delivery. Although some studies report a modest correlation, others find cord blood acidosis to be independent of umbilical artery systolic/diastolic ratios.
The behavior of the fetal autonomic nervous system, as manifest in a variety of patterns of electronically recorded fetal heart rate, have been the mainstay of obstetrical surveillance both before and during labor for the past two decades. At best, these patterns can be viewed as occasional predictors of acute perinatal asphyxia, but they are severely limited by the high frequency (as high as 30 percent) of abnormal patterns in the non-asphyxiated population.1 Blood gases obtained antepartum by cordocentesis have been predicted reasonably well by antepartum fetal heart rate abnormalities, albeit in small, highly-selected populations of fetuses at risk. Less consistent results have been found for associations with cord pH at delivery. Intrapartum fetal heart rate abnormalities correlate so modestly with concurrent pH that many obstetrical authorities advise obtaining capillary pH before intervening in labor on the basis of fetal heart rate abnormalities.
The paper by Dr. Hauth brings us closer to the thing itself. Hauth makes a convincing case from his own work and that of others that the combination of severely depressed pH in the umbilical artery at birth (< 7.0) and an Apgar score of 3 or less at 5 minutes, is a viable definition of what we mean by acute perinatal asphyxia.
Let us examine the rationale for this viewpoint by describing the characteristics of an ideal measure of acute perinatal asphyxia. I see the following four conditions as essential characteristics of this ideal measure:
- It directly reflects the metabolic process we understand by asphyxia, namely interference of gas exchange in the fetoplacental unit;
- It can be obtained reasonably easily in most clinical settings;
- It correlates strongly with the acute neurologic insults characteristically found in those rare infants who have experienced difficult labors and who later have neurological handicap; and
- It correlates strongly with acute neonatal impairment of renal, cardiac or pulmonary functioning.
The physiologic rationale for using severe metabolic acidosis as an indicator of asphyxia is well laid out by Dr. Hauth, satisfying the first condition. The ease of testing for pH on a cord blood specimen immediately after birth and obtaining the Apgar score components satisfies the second condition. Dr. Hauth's review of the recent literature which links very low pH combined with low Apgar score to acute neurological insults and to respiratory insufficiency is convincing enough to satisfy the third and fourth conditions listed above.
It is interesting to note that low pH alone does not satisfy the latter two criteria, but that an indicator of birth depression is needed in order to provide a strong correlation with neonatal conditions. One interpretation of this observation is that birth depression signifies prolonged duration of acidosis. Babies with severe cord blood acidosis without birth depression have acidosis of very recent onset from which they can quickly recover. Another interpretation is that some fetuses are unusually resistant to the effects of asphyxia, as manifest in their lack of birth depression, and are therefore likely to escape their asphyxial insult uninjured.
It may be argued that Hauth's definition of acute perinatal asphyxia is too severe, that it may not include all infants who have suffered acute perinatal asphyxia. The combination of a cord pH < 7.0 and a 5-minute Apgar score £ 3 is indeed rare. In one large study cited by Hauth,2 this combination occurred only five times in a population of 2,738 term deliveries (1.8 per thousand deliveries). But cerebral palsy is found in two to three per 1,000 live births, and between 10 percent (Hauth's estimate) and 25 percent (my own estimate)3 are judged attributable to acute perinatal asphyxia in term infants (0.2 to 0.75 per 1,000). Thus only a minority of infants with such severe acidosis and birth depression will develop cerebral palsy, and less severe grades of acute perinatal asphyxia would presumably be at lower risk.
One inherent limitation of Hauth's definition is that it is static rather than dynamic. The acute in acute perinatal asphyxia implies a time dimension that cannot fully be appreciated by any measurement at a single point in time. When a baby is born asphyxiated we tend to assume that the metabolic changes, as well as the neurologic dysfunction, are of relatively recent onset, and that they did not exist prior to the onset of labor. But how do we know this? It would be intriguing to apply tests of fetal neurobehavioral function (such as are incorporated in the several fetal biophysical profiles) to a large number of fetuses at the onset of labor and compare these to similar observations made in the same infants in the neonatal period. We might perhaps find that some of our infants with hypoxic-ischemic encephalopathy were behaving quite similarly before the onset of labor.
Epidemiologic and clinical research studies cannot proceed without having at their starting point a working case definition. If we accept Dr. Hauth's proposal that the specific combination of low pH and low Apgar described in his paper constitute the working case definition of acute perinatal asphyxia, what research would flow from this?
In the first place, a clear and easily measurable case definition would allow us to obtain a descriptive epidemiology of acute perinatal asphyxia. In a large prospective study, we could observe the relationships of any of the maternal and placental variables described by Drs. Salafia and Low to this case definition, and describe their sensitivity and specificity for the diagnosis of acute perinatal asphyxia.
Such research would also enable us to better contextualize acute perinatal asphyxia in the real world. We have considerable information about the types of pregnancies that are susceptible to acute perinatal asphyxia (admittedly based on less rigorous case definitions than proposed here), but we have little information on the characteristics of the mothers (or for that matter, fathers) whose fetuses are at risk, particularly when the pregnancy presents no special problems. Investigation of whether the incidence of acute perinatal asphyxia varies by season, ethnicity social class, geographic location, or type of obstetrical care would be feasible. Case-control studies could further elucidate the epidemiology of acute perinatal asphyxia, perhaps with more efficiency.
In the second place, it would allow us to follow a cohort of infants classified by their exposure to acute perinatal asphyxia and to determine the risk of later neurological deficits, and how these risks are mediated by neonatal dysfunctions, particularly of the central nervous system. Of particular interest would be to ascertain whether infants with lesser degrees of birth asphyxia contribute to the pool of handicapped children, and the proportion of all cerebral palsy that is attributable to acute perinatal asphyxia.
References
- Steer PJ, Eigbe F. Lissauer TJ et al. Interrelationships among abnormal cardiotocograms in labor, meconium staining of the amniotic fluid, arterial cord blood pH, and Apgar scores. Obstet Gynecol 74:715-721,1989.
- Gilstrap LC, Leveno KJ, Burris J et al. Diagnosis of birth asphyxia on the basis of fetal pH, Apgar score and newborn cerebral dys function. Am J Obstet Gynecol 161:825-830, 1989.
- Paneth N. The etiology of cerebral palsy. Pediatric Ann 15:191-201, 1986.
Discussion
Julian T Parer, M.D., Ph.D.
Department of OB/GYN, University of California, San Francisco
This session covered five areas of fetal assessment:
- fetal heart rate monitoring;
- Doppler velocimetry;
- fetal blood sampling;
- umbilical cord blood sampling; and
- placental pathology.
Each of the authors discussed these areas based on the available literature, and summarized their personal opinions of the utility of the techniques.
I would like to discuss the topic of clinical obstetric assessment with reference to four topics not directly addressed by the speakers:
- What is asphyxia?
- FHR monitoring—what is abnormal?
- What is fetal distress?
- Placental pathology and the clinical assessment of fetal asphyxia.
What Is Asphyxia?
I have noted in my formal presentation in this conference that asphyxia is best defined as insufficiency of exchange of the respiratory gases. Clinically this is almost always because of insufficient umbilical or uterine blood flow, and will result in a reduction in oxygen content and elevation in CO2 tension in fetal blood. Occasionally asphyxia may be the result of reduced maternal arterial oxygen content. Eventually the insufficiency of oxygen will result in a series of physiologic compensatory mechanisms within the fetus, the most important of which is probably redistribution of blood flow within the fetus, resulting in increased blood flow to the heart, brain and adrenal gland, maintenance of blood flow to the placenta (i.e., umbilical blood flow) and reduction in blood flow to essentially all other regions and organs of the body. In those regions where blood flow is reduced, oxygen delivery will be insufficient to maintain oxygen consumption and high energy bond production will occur by means of anaerobic metabolism. The result of this will be initially local tissue or organ lactate production, and eventually a systemic lactic acidemia. As the oxygen supply is reduced further there will be interference with these compensatory mechanisms, such that myocardial performance is affected, and cardiac output is reduced. This will result in a reduction of umbilical blood flow and further aggravation of the asphyxia because of poor oxygen pickup in the fetal placenta. Eventually, the fetus will suffer regional hypoxia to the extent that there is cellular damage.
It is obvious from the above discussion that asphyxia is not a sudden state of being, but rather represents a continuum of insufficiency of exchange of the respiratory gases. This continuum begins as a physiologic asphyxia when we consider the mild mixed metabolic and respiratory acidosis in umbilical cord blood specimens at the time of delivery in essentially all fetuses. Typical values (mean ± SD) are pH 7.2 ± 0.08 PCO2 48 mm Hg and base excess -9 ± 4mEq/l.1 The other end of the spectrum is the extremely severe metabolic and respiratory acidosis seen in babies suffering asphyxial brain damage following a catastrophic event such as a compressed prolapsed cord or complete abruption. In the latter case values would be of the order of pH 6.8, PCO2 90 mm Hg and base excess -20 mEq/l.2
In view of the fact that the state of asphyxia spans a continuum from physiologic to terminal, and that pathologic results such as brain damage or death have not been rigidly defined in terms of acid-base state, it does not seem reasonable to assign any particular value or set of values to the term asphyxia as has been proposed in Dr. Hauth's paper. As he notes the duration and extent of metabolic acidosis and hypoxia that will result in neurologic damage to the human fetus are not known, and that is probably because there is an extremely wide variation of severity and duration of asphyxia that ultimately results in damage. In addition, several other factors may be involved: stage of fetal development, metabolic state (e.g., carbohydrate reserves), and repetitiveness of asphyxial insults.3
An approach taken by Low and coworkers4 was the combination of severity and duration resulting in motor and cognitive defects in approximately 50 percent of children; they defined this as an episode of hypoxia in excess of one hour resulting in a metabolic acidosis of the order of 25 mEq/l (defined by buffer base). This is an appropriate approach to the problem but undoubtedly is of limited predictive utility. The use of specific cutoff values as proposed by Gilstrap and colleagues5 of PCO2 and base deficit in the face of umbilical arterial pH less than 7.20 is, I believe, of limited usefulness. The splitting up into respiratory or metabolic acidemia is artificial and does not apply to the physiologic situation where there is almost invariably a mixed respiratory and metabolic acidosis.
I therefore believe that the term asphyxia should not be used to denote a specific pathologic state but rather should be used to define simply what it is, that is, elevated CO2 and reduced 02, with usually a metabolic component. The term should simply be used as a preceding descriptor for the acid-base features of umbilical arterial blood and the weakness of these values as a predictor of fetal damage should be implied.
In like manner the use of a specific pH value in fetal blood samples as an indication for immediate delivery, such as 7.20, has been discarded by many for over a decade.6 The use of fetal heart rate monitoring as the screen, and fetal blood sampling to determine acid-base status in selected cases is well established by the vast majority of practitioners in North America. As Dr. Hauth points out it is now widely accepted that fetal blood sampling has limited usefulness in intrapartum management, and that usefulness, we believe, is limited to unusual or puzzling heart rate patterns, or those with reduced or absent variability where one is attempting to rule out asphyxia as a cause. At most this should be required in 1 percent of labors, and this means that the use of fetal blood sampling will eventually diminish to zero because of the problems of disuse atrophy .
Fetal Heart Rate Monitoring—What Is Abnormal?
Interpretation of fetal heart rate monitoring patterns is in an extraordinarily confused state in North America, and there is great variation in opinions amongst the 759 certified perinatologists with regard to what is abnormal. There is no argument anymore about the fact that many babies exhibiting various types of decelerations will not be born asphyxiated, yet many people persist in calling such patterns abnormal or some other similar qualitative terms such as nonreassuring or ominous. It is my belief that all of these terms have lost their usefulness and that they should be replaced by other more descriptive terms.
There is general agreement amongst essentially all perinatologists that the baby with a heart rate in the normal range, that is 120-160 bpm, with normal fetal heart rate variability, that is, an amplitude range of 6 bpm or greater, and absent decelerations is essentially always nonasphyxiated. Therefore it seems reasonable to refer to this pattern as the normal fetal heart rate pattern.
There is also general agreement that the fetus with absent fetal heart rate variability, a heart rate within or outside the normal range, and deep variable or late decelerations, is presumed to be asphyxiated, and expeditious delivery is indicated. The same is true of a deep persistent bradycardia, for example 60 bpm or less, with absent variability (in the absence of congenital heart block). Clinically this also represents a fetus with presumed asphyxia and expeditious delivery is indicated.
Between these two extremes, that is, the normal and the presumed asphyxiated baby, there exists a vast array of fetal heart rate patterns which fit neither of these fairly accurate predictions. Thus, the fetus with normal fetal heart rate variability, even in the face of reflex late decelerations or variable decelerations, is almost invariably born nonasphyxiated and vigorous. Such babies should be described as having variant fetal heart rate patterns rather than abnormal fetal heart rate patterns. It is inappropriate to label such patterns fetaldistress because such babies are not asphyxiated, although they may have mild elevations of CO2 and mild depressions of base excess as a result of a cumulative mild asphyxia. Appropriate management for such fetuses is an attempt to abolish the decelerations, or, if unsuccessful, then observation of the evolution of the pattern to be sure that a cumulative asphyxia does not develop resulting in fetal decompensation. This is currently noted by continued retention of normal fetal heart rate variability. Should FHR variability decrease to the point where it can be predicted to result in asphyxial decompensation before a vaginal delivery, then this can be labeled fetal intolerance of labor. This then would be an appropriate indication for operative delivery.
What Is Fetal Distress?
We have previously written at length on the concept of fetal distress,7and the fact that the term is so ill-defined and loosely used that it has lost its usefulness. I believe that an appropriate, though awkward, definition of fetal distress is:
- Progressive persistent asphyxia of such severity that if it is not relieved will result in:
- breakdown of the fetal compensatory blood flow redistribution;
- reduced cerebral metabolism; and
- neuronal cell death.
It is currently widely accepted that this cardiorespiratory decompensation can be clinically noted during labor by reduced or absent fetal heart rate variability in the appropriate setting, that is, in the setting of severe variable or late decelerations, or a bradycardia.
As noted above, in specific cases the term fetal distress can be replaced by more descriptive terms such as presumed fetal asphyxia or fetal intolerance of labor.
Placental Pathology and the Clinical Assessment of Fetal Asphyxia
As a practicing obstetrician, it has been my observation that placental pathology is rarely of utility in determining the etiology of fetal asphyxia. This may not hold true when dealing with certain pathologists who have made a specific practice of specializing in the placenta. However, the vast majority of babies are not born in institutions where such is the case. Perhaps the reason for this is because of the fact that causes of fetal asphyxia tend to be functional rather than anatomic. That is, even in the case of an infarcted fibrotic placenta the adequacy of uterine blood flow cannot be determined simply by pathologic examination, although certainly a small infarcted placenta will support the suspicion that the fetus born asphyxiated and growth retarded is so because of this. However, the functional adequacy of uterine blood flow can only be implied.
A further observation is that the placentas tend to be more carefully examined in cases of abnormal outcome, and true measures of sensitivity and specificity of the abnormal findings cannot be determined from this. It is therefore easy for us to be told that an association is, in fact, a causation. I therefore believe that the maximum utility will be obtained by correlation of pathologic examination with physiologic determinations, and randomized examination of all placentas to determine the actual significance of abnormal findings.
References
- Sykes GS, Johnson P, Ashworth F et al. Do Apgar scores indicate asphyxia? Lancet 1:494-496, 1982.
- Goodwin TM, Belai I, Hernandez P et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol 162:1506-1512, 1992.
- Gluckman PD, Williams CE. When and why do brain cells die? Dev Med Child Neur 34:1010-1021, 1992.
- Low JA, Galbraith RS, Muir DW et al. Factors associated with motor and cognitive deficits in children after intrapartum fetal hypoxia. Am J Obstet Gynecol 148:533, 1984.
- Gilstrap LC, Leveno JK, Burns J et al. Diagnosis of birth asphyxia on the basis of fetal pH, Apgar score, and newborn cerebral dysfunction. Am J Obstet Gynecol 161:825-830, 1989.
- Parer JT. The current role of intrapartum fetal blood sampling. In: Clinical Obstetrics and Gynecology. Resnick R (ed). Harper and Row, 23:565-582, 1980.
- Parer JT, Livingstone EG. What is fetal distress? Am J Obstet Gynecol 162:1421.-1427, 1990.
Session III: Clinical Assessment-- Neonatal
Moderator: Linda L. Wright
Neuroimaging of Perinatal Asphyxia in Term Infants
Marvin D. Nelson, Jr., M.D.
Department of Radiology, Children's Hospital
of Los Angeles, California
Normal Neonatal Brain
Growth and Histologic Appearance
The neonatal brain differs greatly from the adult brain. At birth, the average brain weighs 365 gm., approximately one-fourth the weight of an adult brain.1 All nuclei and fiber tracts are present. Neurons have achieved their maximum number and generally have completed migration. Postnatal growth of the brain represents the production of additional axons, dendrites, astrocytes, supporting cells, and myelin. Primary and secondary sulcation are complete, with tertiary sulcation well under way.2 All cranial nerves are myelinated at birth.3 Myelin is present throughout the spinal cord, medulla, pons, and cerebellum. In the cerebrum, myelin is found in the posterior limb of the internal capsule and in the corona radiata around the central sulcus. Myelination is beginning in the optic tracts, optic chasm, and optic radiations.3 Arachnoid villi are present over the convexity of the brain; however, pacchionian granulations do not develop until the child is approximately 18 months of age.4 The total volume of cerebrospinal fluid (CSF) present at birth averages 50 cc. The amount of CSF produced averages 20 cc/hour. The neonatal brain is 80 to 90 percent water, with little difference between gray and white matter (Table 1).5 This similarity in water content makes it difficult to identify the boundaries of gray and white matter by all imaging methods.
Neuroimaging Apprearance of the Normal Neonatal Brain
Cranial neurosonography uses the property of sound waves to reconstruct an image. Echos are produced when objects of different density produce an interface which causes the sound beam to reflect back to the transducer. In the normal neonatal brain there is little difference in density between gray and white matter, so that little if any discrimination is possible. However, the contours of the external surface of the brain and the size and shape of the lateral ventricles are easily discerned. Following a vaginal delivery, the lateral ventricles may normally appear to be slit-like for 24 to 36 hours after birth. After 36 hours, the ventricles should be large enough so that the walls do not touch each other.
Computed tomography uses x-rays to determine the density of brain tissue to produce a computer reconstructed image of the brain. A CT scan of the normal neonatal brain shows patchy areas of low density through the white matter which may appear to extend throughout the cortex. Again the contours of the surfaces of the brain and size of the ventricles are easily determined. The presence of hemorrhage and calcification is easily determined.
Magnetic Resonance Imaging utilizes the properties of water within the brain tissue to produce a computer reconstructed image of the brain. The different sequences depict how the water molecules interact with their surrounding environment. Since the neonatal brain is mostly water, poor gray/white differentiation is seen and many abnormalities may be hidden in the unmyelinated white matter.
TABLE 1: Composition of the Brain at Different Ages*
*Modified from Folch-Pi5
| AGE | TOTAL SOLID (%) | TOTAL WATER (%) |
|---|---|---|
| Cortex Fetus 3-9 months | 10.40 | 89.6 |
| Cortex Child 2-3 months | 11.0 | 89.0 |
| Cortex Child 2-5 years | 13.0 | 87.0 |
| Cortex Child 11-19 years | 15.7 | 84.3 |
| White Matter Fetus 3-9 months | 9.5 | 90.5 |
| White Matter Child 2-3 months | 15.2 | 84.8 |
| White Matter Child 2-5 years | 25.7 | 74.3 |
| White Matter Child 11-19 years | 30.6 | 69.4 |
Serial (daily) cranial ultrasounds are recommended to evaluate the acutely damaged neonatal brain. Once the child is stabilized, a CT scan of the brain can be obtained to evaluate extent of injury, calcifications and hemorrhages. Magnetic resonance imaging is best used as a follow-up examination when the child is older and more of the white matter has been myelinated.
The Damaged Neonatal Brain
Growth and Histologic Sequential Changes of Damaged Brain Tissue
Both by growth and histologic examination, brain tissue may appear normal for up to twelve hours following an insult. However, by twelve hours, cerebral edema is usually apparent, as is capillary congestion and microscopically red neurons, which suggest irreversible neuronal damage. By 24 to 48 hours, maximum cerebral edema occurs. By 36 to 48 hours, hypertrophic astrocystes and macrophages are present. The myelin begins to disintegrate. In 4 to 7 days, glial fibriles appear within astrocytes. By 7 days, most of cerebral edema has resolved, and there is gross evidence that necrotic tissue has been removed. In 2 to 4 weeks, most of the necrotic tissue has been removed, mineralized neurons may be seen, and chunks of calcification may be present in the cerebral tissue.6
Neuroimaging Appearance of Sequential Changes in the Brain
By cranial ultrasound, CT and MRI, the acutely injured brain may appear normal for the first 12 to 24 hours. The first change by neuroimaging appears to be swelling of the brain, i.e., mass effect resulting from cerebral edema. This may be seen as early as 12 hours, but becomes evident by 24 to 48 hours. Cerebral swelling by all imaging methods appears as mass effect, that is obliteration of sulci, compression of lateral ventricles, loss of cisterns around the brain, brain stem, and cerebellum. By cranial ultrasound, cerebral edema is thought to cause diffuse increased echogenicity throughout the brain tissue.7 By CT, diffuse cerebral edema produces a ground glass appearance with lower attenuation of the entire affected brain tissue.8 By MRI, cerebral edema should produce an increase in intermediate and T2 signals throughout the area affected. However, since the neonatal brain is mostly water, it is very hard to assess any signal change in the affected areas. The increase in signal may only be apparent in areas that have already myelinated. Assuming no further insult, the mass effect begins to reside in 3 to 4 days. Most of the mass effect from cerebral edema is gone by 1 week.
Evidence of non-reversible damage of brain tissue may be seen as early as 24 hours, but typically appears around 72 hours following the insult. By cranial ultrasound, this appears as areas of increased echogenicity within the brain tissue.7 By CT this appears as areas of low attenuation, which is very difficult to distinguish from the surrounding edema.8 By MRI, the damaged area should produce increase in intermediate and T2 weighted signal and decrease in T1 weighted signal, which again would be extremely difficult to distinguish from the surrounding edema and normal brain at this age.9
The first evidence of cavitation within necrotic tissue is usually apparent at seven days following the insult. Again, by ultrasound this appears as a hypoechoic area, i.e., a cystic area within the hyperechoic area of damage. By CT this appears as a CSF density area within the brain tissue, and by MRI again, this should be an area which seems to track CSF. After several months, all of the necrotic tissue should be removed. There may be secondary enlargement of the lateral ventricles to compensate for the loss of brain tissue. By cranial ultrasound, the damaged brain tissue should already be removed and should have the appearance of CSF. The margins of the damaged tissue may remain hyperechoic permanently. By CT the old areas of infarction appear as loss of brain tissue with secondary enlargement of the lateral ventricles. By MRI there is a similar appearance of loss of brain tissue with the secondary enlargement of lateral ventricles. If intravenous contrast is given either by CT or MRI, enhancement may appear within the area of damage as early as 24 hours, but usually the peak enhancement occurs at 2 weeks and enhancement may occur for at least 8 weeks following the insult.10-12 At least 20 percent of infarcts never enhance with contrast.10-12 After 1 to 2 months, calcifications may appear in necrotic brain tissue. These may appear as an hyperechoic focus with distal shadowing by cranial ultrasound, but are best identified by CT. Magnetic Resonance Imaging will not visualize small parenchymal calcifications.
What Can Neuroimaging Tell Us?
Timing the Onset of the Insult
Often the critical determining factor becomes the presence or absence of edema on initial imaging studies. This is usually made by cranial ultrasound and is usually determined by the size of the lateral ventricles. Following a vaginal delivery, slit-like lateral ventricles may be normal for 24 to 36 hours following birth. However, slit-like ventricles beyond this time period are distinctly abnormal and should suggest cerebral edema. Necrotic brain tissue usually becomes hyperechoic between 2 to 4 days following the insult, and shows areas of cavitation within the hyperechoic area by approximately 7 days.
The Pattern of Injury
The pattern of injuries may suggest an etiology. A normal term neonate compromised during labor and delivery shows low profusion injury to the brain from reduced cardiac output. This is evident first by areas of infarction at the parietal vertex and then in the border zones between anterior and middle cerebral arteries and between middle and posterior cerebral artery territories. Similarly, areas in the end distribution of the penetrating vessels at the base of the brain, the striatal and thalamoperforating vessels may be compromised and show areas of necrosis in the superior aspects of the putamen, caudate and thalamus. Collectively, these are known as the vascular border zone areas (watershed). There is no vascular border zone in the white matter surrounding the ventricles in the preterm or term infant.13,14
Focal areas of coagulation necrosis in the white matter (periventricular leucomalacia) suggest maternal and/or neonatal sepsis.15,16
References
- McLennan JE, Gilles FH, Neff R. A model of growth of the human fetal brain. In: Gilles FH, Leviton A, Dooling EC (eds). The Developing Human Brain. Boston: John Wright & PSG, pp. 43-58, 1983.
- Chi JG, Dooling EC, Gilles FH. Gyral development of the human brain. Ann Neurol 1:86-93, 1988.
- Brody B, Kinney H, Kloman A, Gilles E. Sequence of central nervous system myelination in human infancy I. An autopsy study of myelination. J Neuropathol Exp Neurol 46:283-301, 1987.
- Gilles FH, Kassirer M. Hydrocephalus. Hum Pathol 7:46:283-301, 1976.
- Folch-Pi J. Composition of the brain in relation to maturation. In: Waelsch H (ed). Biochemistry of the Developing Nervous System. New York: Academic Press, pp. 121-132, 1955.
- Garcia JH. Circulatory disorders and their effect on the brain. In: Davis RL, Robertson DM (eds). Textbook of Neuropathology. Baltimore: Williams and Wilkins. Chapter 12, 1985.
- Siegel MJ, Shackelford GD, Perlman JM, Fulling KH. Hypoxic-ischemic encephalopathy in twin infants: Diagnosis and prognosis evaluated by ultrasound. Radiol 152:395-399, 1984.
- Davis KR, Taveras JM, New PJF et al. Cerebral infarction diagnosis by CT. Am J Roentgenol 124:643-660, 1975.
- Johanson MR, Pennoek JM, Bydder GM et al. Serial MR imaging in neonatal cerebral injury. AJNR 8:83-92, 1984.
- Norton GA, Kishore PRS, Lin J. CT contrast enhancement in cerebral infarction. Am J Roentgenol 131:881-885, 1978.
- Wing SD, Norman D, Pollack JA, Newton TH. Contrast enhancement of cerebral infarcts in CT. Radiol 121:89-92, 1976.
- York DH, Jr, Marshall WH, Jr. Recent ischemic brain infarcts at computed tomography: Appearance pre- and post-contrast infusion. Radiol 117:599-608, 1975.
- Nelson MD, Jr, Gonzalez-Gomez I, Gilles FH. The search for human telencephalic ventriculfugal arteries. AJNR 12:215-222, 1991.
- Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: An anatomic study. Am J Neuroradiol 11:431-439, 1990.
- Leviton A, Paneth N. White matter damage in preterm newborns—an epidemiologic perspective. Early Human Dev 24:122, 1990.
- Gilles FH, Averill DR, Kerr CS. Neonatal endotoxin encephalopathy. Ann Neurol 2:48-56, 1977.
Differential Diagnosis and Contribution of EEG
Robert R. Clancy, M.D.
Professor of Neurology and Pediatrics
The University of Pennsylvania School of Medicine
and The Children's Hospital of Philadelphia
The Differential Diagnosis of Neonatal Encephalopathy
Neonatal encephalopathy may be broadly recognized by the presence of clinical abnormalities of neurologic function including:
- altered mental status (i.e., lethargy or coma);
- seizures;
- neuromuscular signs (inactivity, hypo/ hypertonia, hypo/hyperreflexia); and
- bulbar dysfunction (impaired suck or swallow, apnea, etc.).
Neonatal encephalopathy may arise from acute disorders that generally provoke acute electroencephalogram (EEG) changes (Table 1). Chronic disorders such as cerebral dysgenesis are associated with long standing or chronic EEG abnormalities. Peripheral nervous system disorders such as Werdnig-Hoffman disease may mimic an encephalopathy by the presence of bulbar and neuromuscular signs but preserved mental status and normal EEG (Table 1).
EEG Is a Sensitive But Nonspecific Measure of Brain Function
The generators of the neonatal EEG signal are not exactly known but are presumed to reside in the cerebral cortex which is regulated by the modulating influence of the thalami, reticular activating system and upper brainstem. Thus, the EEG is a reflection of whole brain function.
Any medical or neurological condition that affects brain function may alter the EEG. If anything, the EEG is too sensitive since even mild, transient and non-injurious factors (e.g., mild hyponatremia) may induce visible EEG changes. ln such circumstances, abnormal EEGs are not equivalent to a damaged central nervous system (CNS) but rather to a temporary and reversible malfunction (Figure 1).
TABLE 1: Differential Diagnosis of Conditions Which Appear as or Mimic Neonatal Enchephalopathy
HIE = Hypoxic-ischemic encephalopathy
NS = Neonatal seizures
EMEE = Early myoclonic epileptic encephalopathy
EIEE = Early infantile epileptic encephalopathy (Ohtanara's syndrome)
See References 2, 5, 6, 9, 16, 18, 20, 21, 23, 29, 31-34
| CENTER | EEG CHANGE |
|---|---|
| ACUTE CEREBROVASCULAR | acute |
| ACUTE CEREBROVASCULAR | acute |
| ACUTE CEREBROVASCULAR | acute |
| ACUTE CEREBROVASCULAR | acute |
| TOXIC/METABOLIC | acute |
| EPILEPTIC benign NS, EMEE, EIEE | acute |
| CHRONIC CNS in utero injury | chronic or normal |
| CHRONIC CNS brainstem dysfunction | chronic or normal |
| CHRONIC CNS cerebral dysgenesis | chronic or normal |
| Werdnig-Hoffman | normal |
| Gillian Barre Syndrome | normal |
| Myastenia gravis | normal |
| Congenital myopathy | normal |
| Myotonic dystrophy | normal |
| Familial dysautonomia | normal |
Abnormal EEGs are not specific from the viewpoint of etiology. A burst suppression EEG may arise from a severe acute encephalopathy due to global hypoxia-ischemia, intracranial hemorrhage or overwhelming infection.
FIGURE 1: Evolution of EEG Changes During and After an Acute Perturbation
Measurement of EEG Changes and Abnormalities
Visual Analysis
EEGs are traditionally interpreted by visual analysis of the tracing. The EEG background is the fundamental stage (the ongoing, moment-to- moment cerebral electrical activity) upon which the brief appearance of transient events (electrographic seizures, sharp waves) may impose. The background is judged by its:
- continuity;
- symmetry;
- interhemispheric synchrony;
- voltage; and
- composition of specific patterns and rhythms that are expected at a given conceptional age (CA).
Semi-quantitative methods have evolved to describe EEG abnormalities such as excessive discontinuity asymmetries, interhemispheric asynchrony, low voltage or isoelectricicy and paucity of the normal background pattems for age.
FIGURE 2: EEG Discontinuity
Continuity is a relatively easily quantified parameter of the EEG background. In the discontinuous segments of an EEG, periods or bursts of activities (B1, B2, B3...) are interrupted by lower voltage quiescent periods (Q1, Q2,Q3 ....) (Figure 2). The duration of the quiescent or interburst interval (IBI) is sensitive to brain maturity (conceptional age) and the presence of medical or neurologic illness.15
Typically, the IBI increases, sometimes extremely so, in the presence of disease. This resembles a regression to a more primitive or immature state in which the IBIs are characteristically lengthened.
Most clinical classification schemes distill the interpretation of the EEG background into one of the following categories:
- normal or mildly abnormal;
- moderately abnormal; or
- markedly abnormal.
An example of one scheme that has been empirically validated is provided as Table 2.
TABLE 2: Classification of EEG Background Activity
|
1. MILDLY ABNORMAL
|
|
2. MODERATELY ABNORMAL
|
|
3. MARKEDLY ABNORMAL
|
Computer Assisted Quantification
Cerebral function monitors can electronically and mathematically decompose a raw EEG signal and convert complex analog waveforms into a digital format.3,4,17,19 Commonly, the digital signal is epresented by the power of the EEG (picowatts) expressed in frequency bins such as the familiar beta, alpha, theta and delta bandwidths. Each brain region of interest can be depicted as a mean + standard deviation value for power at each frequency bin. Normative data have been generated for different conceptional ages and characteristic alterations with diseases have been observed. A prime advantage of this technique is the quantification of physiologic data and avoidance of subjective interpretive error by inexperienced EEG readers.
EEG Changes and Abnormalities Parallel Intercurrent Illness
The basic working principal of clinical neonatal EEG interpretation is that there should be a reasonable proportionality between the severity of an illness and its impact on cerebral function and consequently the EEG. A mild illness is reasonably expected to incite a mild EEG disturbance whereas an overwhelming cause of encephalopathy will usually produce a severe EEG disturbance.25 For example, Tharp (Figure 3) reported that the duration of the average interburst interval increases with lower oxygen values, after a brief lag time.36
Similarly, in a study of serial EEGs in severely encephalopathic infants with hyperammonemia due to citrullinemia, Clancy and Chung (Figure 4) showed a significant correlation between higher serum ammonia levels, increased intervals and worsening clinical neurological status.12
FIGURE 3: Example of Interburst Interval Changes Due to Systemic O2 Variations
FIGURE 4: Correlation of Serum Ammonia Levels and Interburst Intervals in Three Neonates with Citrullinemia
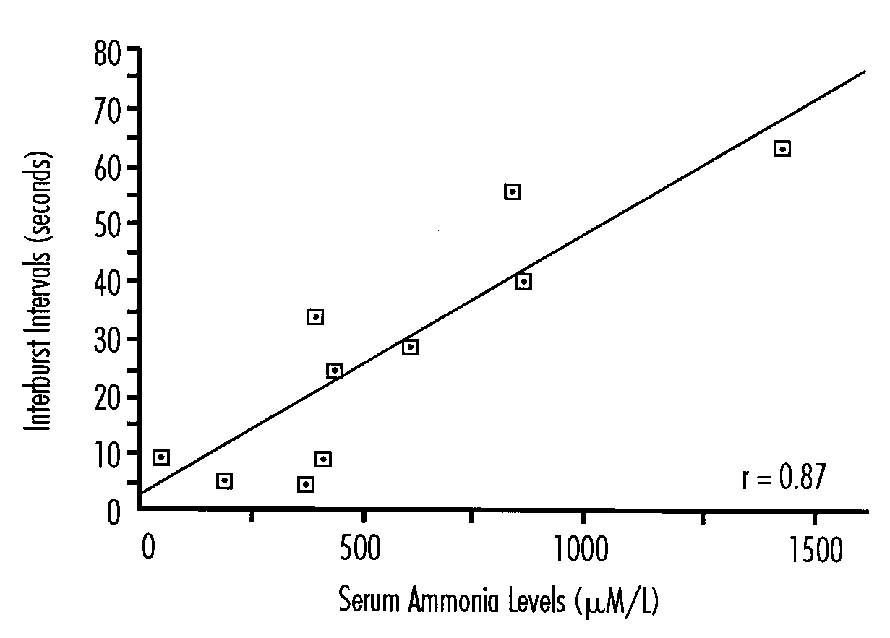
FIGURE 5: Neuropathologic Correlation with EEG Abnormalities
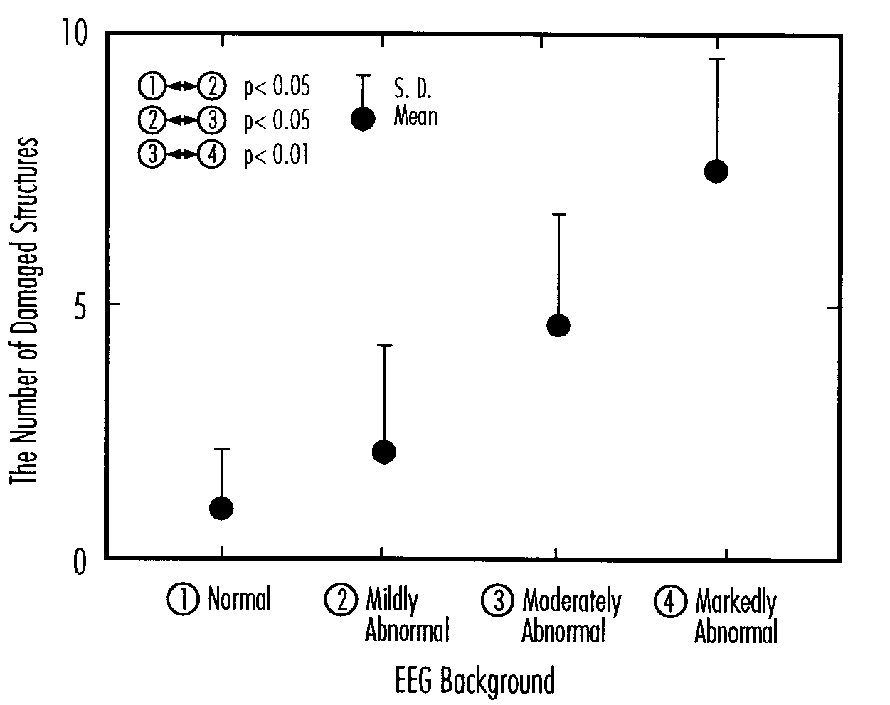
FIGURE 6: Acute Encephalopathy and Evolving EEG Abnormalities
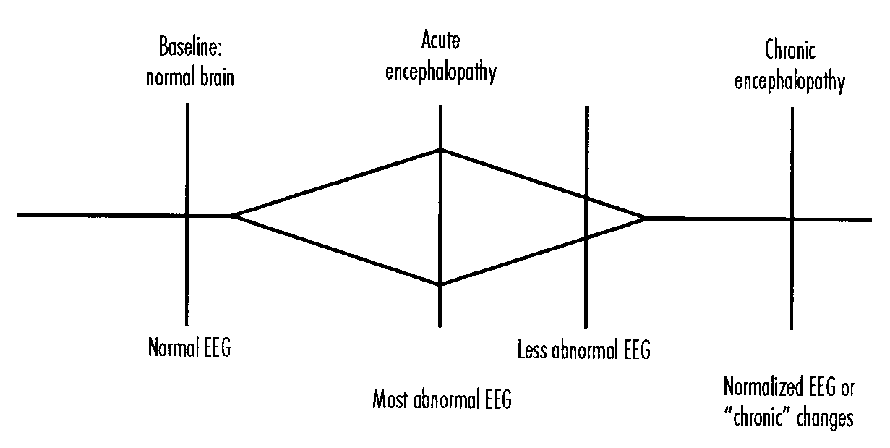
Finally, Aso et al. (Figure 5) reported that the number of abnormal brain structures, determined by postmortem examination, was significantly higher in infants whose EEGs were markedly abnormal compared to those with more normal pre-mortem EEG examinations.1
Temporal Relationship of Encephalopathy to EEG Abnormalities
It is generally recommended that serial EEGs during the height of an illness or encephalopathy should be performed to capture the most abnormal tracing (Figure 6). Basically, this intends to answer the question "how severe was the illness's CNS impact at its worst?" If, at the height of the illness, the EEG was only mildly abnormal, an expectation for a favorable outcome would be justified. However, if the most abnormal tracing was markedly disturbed, such as burst suppression, the conclusion that the illness had likely provoked permanent brain damage is usually justified. Because of the ability of the EEG to non-specifically normalize in the wake of even a severe acute encephalopathy, EEGs obtained long after the inciting event, such as the hospital discharge EEG, frequently lack prognostic predictive power.
It is important to recognize that the EEG can be markedly abnormal for a very brief period of time in special circumstances which do not automatically oblige a poor neurologic outcome. For example, a transient, marked EEG abnormality may occur immediately following a cardiac arrest, seizure or drug infusion such as diazepam (Figure 7). However, EEG abnormalities that persist and endure beyond the insult are more clinically significant (Figure 8).
Aside from the quality of the EEG background, transient EEG phenomena also appear after the onset of an acute insult and gradually dissipate in the days and weeks that follow the resolution of the encephalopathy. Provoked neonatal seizures typically last for a restricted time, then gradually resolve.35 Positive temporal sharp waves are a pathologic EEG transient that may be seen in some term infants who sustain structural brain abnormalities acutely.10,27 They are the mature counterpart to the positive rolandic sharp (PRS) waves and positive vertex sharp (PVS) waves which were first described in premature infants with intraventricular hemorrhage (IVH) or periventricular leukomalacia (PVL).7,11,24 PRS and PVS also appear briefly after the onset of IVH or PVL and then gradually disappear from the background.
Neurologic Prognosis As a Function of the Most Abnormal EEG Background
Numerous published studies have shown the prognostic significance of the most abnormal EEG background.22,26,28 Infants who displayed, at worse, only a mildly abnormal EEG background generally survive their illness and subsequently display robust neurologic development. However, in those who had definite marked EEG abnormalities, there is a much greater chance of death or survival with adverse neurologic outcome. The relationship between EEG background and prognosis has been examined the most carefully in the area of neonatal seizures and intraventricular hemorrhage (Table 3),15,37 but also holds true for other causes of encephalopathy such as meningitis.8
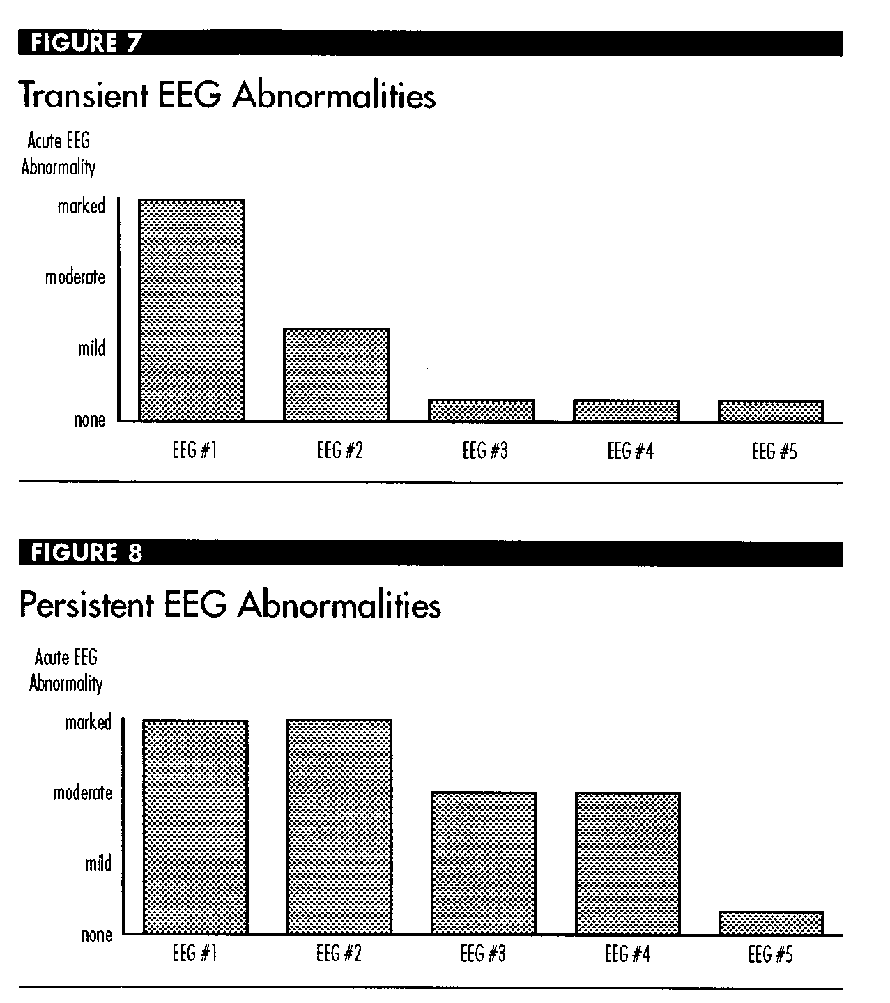
TABLE 3: Outcome Probabilities Predicted by the "Most Abnormal EEG" in Premature Infants with IVH
Model 1: LOGIT (probability of survival) = 3.0161 - 1.6291 ("most abnormal EEG")
Model 2: LOGIT (probability of favorable outcome) = 4.8282 - 3.2188 ("most abnormal EEG")
*P < 0.009
+ P<0.008
From: Clancy et al14
| MOST ABNORMAL EEG | MODEL 1* PROBABILITY OF SURVIVAL | >MODEL 2+ PROBABLITY OF FAVORABLE OUTCOME |
|---|---|---|
| normal/mild | 0.8001 | 0.8333 |
| moderate | 0.4398 | 0.1667 |
| marked | 0.1333 | 0.0079 |
References
- Aso K, Scher MS, Barmada MA. Neonatal electroencephalography and neuropathology. J Clin Neurophysiol 6:103-123, 1989.
- Barron TF, Gusnard DA, Zimmerman RA, Clancy RR. Cerebral venous thrombosis in neonates and children. Pediatr Neurol 8:112-116, 1992.
- Bell AH, McClure BG, Hicks EM. Power spectral analysis of the EEG of term infants following birth asphyxia. Dev Med Child Neurol 32:990- 998, 1990.
- Bell AH, McClure BG, McCullagh PJ, McClelland RJ. Variation in power spectral analysis of the EEG with gestational age. J Clin Neurophysiol 8:312-319, 1991.
- Blair E. A research definition for 'birth asphyxia'? Dev Med Child Neurol 35:449-455, 1993.
- Blair E, Stanley FJ. Intrapartum asphyxia: A rare cause of cerebral palsy. J Pediatr 112:515-519, 1998.
- Blume WT, Dreyfus-Brisac C. Positive rolandic sharp waves in neonatal EEG: Types and significance. Electroencephalog Clin Neurophysiol 53:277-282, 1982.
- Chequer RS, Tharp BR, Dreimane D et al. Prognostic value of EEG in neonatal meningitis: Retrospective study of 29 infants. Pediatr Neurol 8:417-422, 1992.
- Chicayat D, Meagher-Villemure K, Mamer OA et al. Brain dysgenesis and congenital intracerebral calcification associated with 3- hydroxyisobutyric aciduria. J Pediatr 121:86-89, 1992.
- Chung HJ, Clancy RR. Significance of positive temporal sharp waves in the neonatal electroencephalogram. Electroencephalog Clin Neurophysiol 79:256-263, 1991.
- Clancy RR. Interictal sharp EEG transients in neonatal seizures. J Child Neurol 4:30-38, 1989.
- Clancy RR, Chung HJ. EEG changes during recovery from acute severe neonatal citrullinemia. Electroencephalog Clin Neurophysiol 78:222-227, 1991.
- Clancy RR, Tharp BR, Enzman D. EEG in premature infants with intraventricular hemorrhage. Neurology 34:583-590, 1984.
- Connell J, de Vries L, Oozeer R et al. Predictive value of early continuous electroencephalogram monitoring in ventilated preterm infants with intraventricular hemorrhage. Pediatrics 82:327-343, 1988.
- Coorssen EA, Msall ME, Duffy LC. Multiple minor malformations as a marker for prenatal etiology of cerebral palsy. Dev Med Child Neurol 33:730-736, 1991.
- Eyre JA, Nanei S, Wilkinson AR. Quantification of changes in normal neonatal EEGs with gestation from continuous five-day recordings. Dev Med Child Neurol 30:599-607, 1988.
- Govaert P, Vanhaesebrouck P, De Praeter C et al. Moebius sequence and prenatal brainstem ischemia. Pediatrics 84:570-573, 1989.
- Guthrie RD, Knauss TA, Haberkern CM et al. Power spectral analysis of the neonatal primate electroencephalogram during acute hypoxemia. Pediatr Res 16:30-34, 1982.
- Hale DE, Bennett MJ. Fatty acid oxidation disorders: A new class of metabolic diseases. J Pediatr 121:1-11, 1992.
- Holm VA, Cassidy SB, Butler MG et al. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 91:398-402, 1993.
- Holmes G, Rowe J, Hafford J et al. Prognostic value of the electroencephalogram in neonatal asphyxia. Electroencephalog Clin Neurophysiol 53:60-72, 1982.
- Leviton A, Nelson KB. Problems with definitions and classifications of newborn encephalopathy Pediatr Neurol 8:85-90, 1992.
- Marret S, Parain D, Jeannot E et al. Positive rolandic sharp waves in the EEG of the premature newborn: A five year prospective study. Arch Dis Child 67:948-951, 1992.
- McCutchen CB, Coen R, Iragui VJ. Periodic laceralized epileptiform discharges in asphyxiated neonates. Electroencephalog Clin Neurophysiol 61:219-217, 1984.
- Monod N, Pajot N, Guidasci S. The neonatal EEG: Statistical studies and prognostic value in full-term and preterm babies. Electroencephalog Clin Neurophysiol 32:529-544, 1972.
- Nowack WJ, Janati A, Angtuaco T. Positive temporal sharp waves in neonatal EEG. Clin Electroencephalog 20:196-201, 1969.
- Obrecht R, Pollock MA, Evans S, Scott DF. Prediction of outcome in neonates using EEG. Clin Electroencephalog 13:46-49, 1982.
- Roland EH, Hill A, Norman MG et al. Selective brainstem injury in an asphyxiated newborn. Ann Neurol 23:89-92, 1988.
- Ryan SG, Wiznitzer M, Hollman C et al. Benign familial neonatal convulsions: Evidence for clinical and genetic heterogeneity. Ann Neurol 29:469-473, 1991.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress. Arch Neurol 33:696-705, 1976.
- Scher MS, Belfar H, Martin J, Painter MJ. Destructive brain lesions of presumed fetal onset: Antepartum causes of cerebral palsy. Pediatrics 88:898-906, 1991.
- Scher MS, Aso K, Beggarly ME et al. Electrographic seizures in preterm and full-term neonates: Clinical correlates, associated brain lesions, and risk for neurologic sequelae. Pediatrics 91:128-134, 1993.
- Singh I, Johnson GH, Brown FR. Peroxisomal disorders. AJDC 142:1297-1301, 1988.
- Takeuchi T, Watanabe K. The EEG evolution and neurological prognosis of perinatal hypoxia neonates. Brain Dev 11:115-120, 1989.
- Tharp BR. Intensive video/EEG monitoring of neonates. Adv Neurol 46:107-126, 1986.
- Tharp BR, Cukier F, Monod N. The prognostic value of the electroencephalogram in premature infants. Electroencephalog Clin Neurophysiol 51:219-236, 1981.
Neonatal Diagnosis of Perinatal Asphyxia*
Gerald B. Merenstein, M.D.
University of Colorado Health Sciences Center,
The Children's Hospital, Denver
Brian S. Carter, M.D.
Department of Pediatrics, Fitzsimons Army
Medical Center, Aurora, Colorado
Introduction
The terms fetal distress and perinatal asphyxia have become controversial labels rather than straightforward medical diagnoses. The use of ICD-9-CM code 768 intrauterine hypoxia and birth asphyxia, with its subsets 768.1 - 768.6, including fetal distress and moderate or severe birth asphyxia, have added to the confusion. Cooperative efforts of the American Academy of Pediatrics (AAP) Committee on Fetus and Newborn, the American College of Obstetricians and Gynecologists (ACOG) Committee on Obstetrics, the American Hospital Association and the National Center for Health Statistics have recently made changes that will allow the more appropriate use of these codes. Recognizing that the presence of meconium, low Apgar scores, or the use of suctioning and oxygen for babies are not in and of themselves indicative of perinatal asphyxia, terms such as meconium in liqueur, passage of meconium, 1-minute Apgar score of 0-3 and 1-minute Apgar score of 4-7 have all been deleted from the explanation of these terms. In addition, white asphyxia and blue asphyxia have also been deleted. In their joint statement on the use and abuse of the Apgar score the AAP and ACOG have noted that there may be multiple reasons, such as sepsis, prematurity etc., that infants have low Apgar scores and require resuscitation. Therefore the following two descriptions have also been deleted:
- Pulse <100 per minute at birth and falling or steady, respiration absent or gasping, color poor, muscle tone absent;
- Normal respiration not established within one minute, but heart rate =100, some muscle tone present, some response to stimulation.
The ICD-9 descriptions for perinatal asphyxia now read:
- 768 Intrauterine hypoxia and birth asphyxia
Use only when associated with newborn morbidity classifiable elsewhere; - 768.5 Severe birth asphyxia
Birth asphyxia with neurologic involvement; - 768.6 Mild or moderate birth asphyxia
Other specified birth asphyxia (without mention of neurologic involvement).
*This research was supported by grant number M01 RR00069, General Clinical Research Resources, NIH.
Neurologic Involvement
The use of junk science1 in lawsuits has caused the courts to associate fetal distress and perinatal asphyxia with adverse neurodevelopmental outcome despite the fact that less than 10 percent of cases of cerebral palsy or mental retardation are associated with intrapartum perinatal asphyxia.2-6 Central nervous system injury in the newborn may have multiple etiologies including intrapartum hypoxia, intracranial hemorrhage, metabolic disorders (hypoglycemia, hypocalcemia), drug withdrawal, congenital viral infection, acute viral or bacterial infection, neurodevelopmental defects and others.7,8 Central nervous system injury manifests in the term infant with seizures, apnea, respiratory arrest, hyperalertness, jitteriness, posturing, movement disorders, impaired suck, swallow, gag and feeding, hypotonia and abnormal oculomotor and pupillary responses.9 In 1976 Sarnat and Sarnat10 described the syndrome of hypoxic- ischemic encephalopathy (HIE) associated with fetal distress and perinatal asphyxia. The three stages of HIE include Stage I (first 12 to 24 hours): hyperalertness, hyperexcitability, seizures, apnea, jitteriness, weakness; Stage II (24 to 72 hours); coma, respiratory arrest, apnea, abnormal oculomotor reflexes, impaired pupillary response and Stage III (>72 hours); persistent stupor, abnormal suck, swallow and gag, weakness and severe hypotonia.7,11,12
Many authors have used the stages of HIE and EEG findings in the management and prognostication of affected newborns.8,12-14 Although this syndrome is most often associated with perinatal asphyxia, it is clear that there are other etiologies with different timing of the central nervous system insult to be considered.8,12 Nelson and Leviton clearly state that the proportion of HIE due to asphyxia is unknown.8
Multiorgan Effects
A neonate who has had intrapartum hypoxia severe enough to result in HIE will show other signs of hypoxic damage including multiorgan system dysfunction.8,15 This multi-system involvement most likely results from a redistribution of cardiac output away from the skin, muscles, kidneys, lungs, liver and gastrointestinal tract to the heart, brain and adrenal glands.16,17 We have reported the following effects and definitions of non-CNS organ dysfunction in term infants with perinatal asphyxia.11,18
Cardiovascular. Tricuspid insufficiency, myocardial necrosis, shock/hypotension (systolic blood pressure <50 mmHg, need for pressor agents or repeated colloid infusions to maintain blood pressure) and congestive heart failure not associated with structural heart disease or abnormal fluid status.
Gastrointestinal. Necrotizing enterocolitis, hepatic dysfunction—elevated transaminase, prolonged pro-thrombin or partial thromboplastin times with no evidence of disseminated intravascular coagulation.
Hematologic. Thrombocycopenia, disseminated intravascular coagulopathy (DIC).
Metabolic. Metabolic acidosis, hypoglycemia, hypocalcemia, hyponatremia.
Pulmonary. Pulmonary hypertension, surfactant deficiency meconium aspiration—ventilator dependent respiratory distress or hood oxygen requirement for >24 hours.
Renal. Acute tubular necrosis (ATN), oliguria-anuria beyond the first 24 hours, hematuria-proteinuria on successive samples, renal tubular acidosis, hypertension.
In prospectively evaluating a group of infants we noted significantly greater organ dysfunction for infants with severe versus moderate or no asphyxia.18 In severe asphyxia dysfunction of the cardiovascular, central nervous, gastrointestinal, renal and respiratory systems was seen in over 50 percent of the infants. In moderate asphyxia 40 percent of the infants had dysfunction of the cardiovascular and respiratory systems.
Criteria for Neonatal Diagnosis
The program for the 1993 American Pediatric Society/Society for Pediatric Research annual meeting contained several abstracts on perinatal asphyxia.19 Unfortunately there was no consistency in the definition utilized in these studies. Criteria cited included 5-minute Apgar score <7, 5-minute Apgar £ 7, 1-minute Apgar score <7, cord pH <7.15, cord pH <7.20, need for ventilation at resuscitation, gasping at 1 minute, apnea at 1 minute. We have looked at these and similar indicators of asphyxia and additionally, abnormal fetal heart rate tracing, elevated nucleated red blood cell count, and > 1 minute of positive pressure ventilation during resuscitation.18 The correlation of these indicators with multiple organ dysfunction was only significant for 5-minute Apgar score <6, abnormal fetal heart rate tracing and base deficit >10 mEq/L.
A number of laboratory indices of varying sensitivity and specificity have been associated with perinatal asphyxia. These include cerebrospinal fluid, blood or urine lactate, hypoxanthine, pyruvate, creatine phosphokinase and beta 2 microglobulin.20-25
The significance and prognostic value of these tests are unclear.22,25,26 They may have no better predictive value than abnormal Apgar scores or cord pH.25
Acid-base Status
Measurement of umbilical cord or initial neonatal umbilical arterial blood gases and the interpretation of acid base status are important in assessing the effects of perinatal asphyxia. Measurement of arterial gases involves actual values (PaCO2 and pH) and calculations from these (base excess, bicarbonate). Acidemia is defined as an abnormally low pH. Consideration of PaCO2, bicarbonate and base excess help classify the acidemia as metabolic, respiratory or mixed. A low pH simply from an elevated PaCO2, respiratory acidosis, is not usually associated with significant short or long term sequelae.
An umbilical artery pH <7.20 has historically been considered acidosis, but a lower normal range has been suggested by recent studies. Several large epidemiologic studies have suggested values of 7.16, 7.15 or 7.10 as the lower limits of normal.9,27-29 Goodwin et al.30 has reviewed 129 cases in which the umbilical artery pH was less than 7.00 derived from a population of nearly 70,000 term deliveries. Post-asphyxial morbidity (both neurologic and non-neurologic) increased with every 0.10 decrement in pH. No infant with a pure metabolic acidemia was abnormal at discharge. Eleven percent of infants with respiratory acidemia were abnormal but the majority of abnormal infants had a mixed acidemia. In other studies no significant end organ dysfunction was seen in infants with pH>7.00.31,32 A base excess of - 20 mEq/L has been associated with CNS dysfunction.18,33 Unfortunately most studies of fetal/neonatal acid base status do not give values for base excess.
Electronic Fetal Monitoring
Electronic fetal monitoring has become one of the most widely used technologies in obstetrics. Many felt the widespread use of electronic fetal monitoring (EFM) would decrease the incidence of cerebral palsy and mental retardation.34 Prospective trials comparing electronic fetal monitoring to auscultation have not shown differences in perinatal mortality or morbidity, cord blood gases, Apgar scores or long-term outcome.35,36 Freeman has noted that "the hoped-for benefit from intrapartum electronic fetal monitoring has not been realized."37
Apgar Scores
The Apgar score assigned at 1 and 5 minutes of age was designed to assist in the clinical assessment and resuscitation of the newborn, yet it has also been used to diagnose perinatal asphyxia and predict long-term outcome.38-40 Improper and inconsistent scoring affects the Apgar score, as does prematurity, maternal anesthesia and analgesia, congenital infection, and congenital neuromuscular disorders. In addition, clinicians have tended to overestimate the prognostic significance of the Apgar score.41 The AAP, ACOG and the American Hospital Association have established guidelines for the proper use of the Apgar score to avoid the sole use of the Apgar score in diagnosing perinatal asphyxia.15,42,43
Neonatal Diagnosis of Perinatal Asphyxia
In the third edition of Guidelines for Perinatal Care the AAP/ACOG provide the following definition:15
FIGURE 1: Scoring System for Post-Asphyxia Morbidity
"FHR" indicates fetal heart rate
=6 points denotes severe morbidity
| 0 POINTS | 1 POINT | 2 POINTS | 3 POINTS | |
|---|---|---|---|---|
| 5 minute Apgar Base Deficit (mEQ/L) | >6 | 5-6 | 3-4 | 0-2 |
| <10 | 10-14 | 14-19 | =20 | |
| FHR Tracking | Normal | Variable deceleration | Severe variation or late deceleration | prolonged bradycardia |
The term asphyxia should be reserved for a clinical context of damaging acidemia, hypoxia and metabolic acidosis. A neonate who has had hypoxia proximate to delivery severe enough to result in hypoxic encephalopathy will show other evidence of hypoxic damage including all of the following:
- Profound metabolic or mixed acidemia (pH <7.00) on an umbilical cord arterial sample, if obtained;
- Persistence of an Apgar score of 0-3 for longer than 5 minutes;
- Neonatal neurologic sequelae, e.g., seizures, coma, hypotonia; and
- Multiorgan system dysfunction, i.e., cardiovascular, gastrointestinal, hematologic, pulmonary or renal system.
This definition is appropriate but excludes infants without CNS involvement. It also requires observation of the infant for a variable time period which could delay potential therapies. Others have also suggested the use of severe acidemia (pH<7.00) and 5-minute Apgar score of <3.31,32 Combined use of EFM, cord pH and Apgar score has been suggested.44,45
In an analysis of many proposed diagnostic tests (criteria) we found, as mentioned above, significant correlation with multiple organ failure only with graded abnormalities in fetal heart rate tracings up to 2 hours prior to delivery, a 5-minute Apgar score of <6 and base deficit >10 mEq/L. These criteria were weighted and developed into a scoring system using sensitivity testing and chi square analysis. The scoring system is shown below. Severe morbidity, three or more organ system dysfunction, is associated with a score of six or more.
We evaluated the specificity of this scoring system for post-asphyxia morbidity in a large prospective study. Over 3,200 patients (>36 weeks gestation) born in our hospitals were studied, 11 percent of whom required Level II (n=275) or III (n=91) NICU care. Eleven of these patients scored ³ 6 points, and demonstrated significant multiple organ system morbidity: seizures (2); hypoxic-ischemic encephalopathy (5); need for intermittent mandatory ventilation (5); or ³ 24 hours of hood oxygen (4); hypotension (7); hypoglycemia/hypocalcemia (4); DIC/thrombocytopenia (3); and hematuria-proteinuria/oliguria/ATN (9); (Table 2). These 11 patients had a mean ± SD umbilical arterial pH of 6.98 ± 0.18 (range 6.68-7.29) and base deficit of 17.1 ± 6.5 (range 4.0 to 27). The only three deaths in the total population studied were encountered as a) an intrapartum demise (score of 5; autopsy inconclusive), b) an infant dying of lethal cardiac anomalies at <24 hours of age (score of 5), and c) an infant dying with congenital diaphragmatic hernia (score of 6). When compared with a 5-minute Apgar score of <3, umbilical arterial pH cutoff of <7.20, 7.10 or 7.00, and base deficit of >10, an asphyxia score of ³ 6 yielded excellent specificity (0.99) and positive predictive value (0.73) for multiple (³ 3) organ system morbidity (Fisher's Exact Test, p<.001). The negative predictive value for a score ³ 5 was 0.99 but the positive predictive value was only 50 percent.
TABLE 2: Post-Asphyxia Morbidity
| SCORE | >3 ORGAN SYSTEM MORBIDITIES | <2 ORGAN SYSTEM MORBIDITIES |
|---|---|---|
| =6 | 8 | 3 |
| =5 | 23 | 3204 |
| Sensitivity = 0.26 | Chi-square analysis (Fisher's Positive Exact Test) | |
| Specificity = 0.99 | p<.001 | |
| Positive Predictive Value = 0.73 | X2=324 | |
| Negative Predictive Value = 0.99 | Odds Ratio=399 95% CI (59,718) |
The present concerns in clinical and research medicine revolve around both a definition of acute perinatal asphyxia and an ability to identify the infant at greatest risk for morbidity related to such (e.g., predict morbidity). Many of the currently used parameters are too sensitive and end up broadly defining asphyxia in such a way as to imply that it is a frequent event…but without apparent acute morbidity. The scoring system presented here, encompasses both immediate antepartum and postpartum parameters and acid-base status proximate to delivery. It allows a more specific graded definition of asphyxia which is both sensitive and specific for the presence of acute morbidities, and holds a strong enough predictive value to identify patients for aggressive clinical evaluation and management, for referral from community level hospitals to tertiary facilities, for possible new therapeutic interventions, and, perhaps, for long-term follow-up. To date, no data is available on the correlation with this morbidity index and long term outcome.
References
- Huber PW. Galileo's Revenge: Junk Science in the Courtroom. New York: Basic Books, pp. 75-91, 1991.
- Blair E, Stanley FJ. Intrapartum asphyxia: A rare cause of cerebral palsy. J Pediatr 112:515, 1988.
- Committee on Obstetrics: Maternal-Fetal Medicine. Fetal and Neonatal Neurologic Injury, American College of Obstetricians & Gynecologists, Technical Bulletin 163, Washington, DC, ACOG, January 1993.
- Naeye RL, Peters EC, Bartholomew M et al. Origins of cerebral palsy. Am J Dis Child 143:1154, 1989.
- Nelson KB, Ellenberg JH. Antecedents of cerebral palsy: Multivarriate analysis of risk. N Engl J Med 81:315, 1986.
- Paneth N, Stark RI. Cerebral palsy and mental retardation in relation to indicators of perinatal asphyxia: An epidemiologic overview. Am J Obstet Gynecol 147:960, 1983.
- Volpe JJ. Neurology of the Newborn. 2nd edition. Philadelphia: W.B. Saunders, 1987.
- Nelson KB, Leviton A. How much of neonatal encephalopathy is due to birth asphyxia? Am J Dis Child 145:1325, 1991.
- Vintzileos AM, Egan JF, Campbell et al. Asphyxia at birth as determined by cord blood pH measurements in preterm and term gestations: Correlation with neonatal outcome. J Matern-Fetal Med 1:7, 1992.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress. Arch Neurol 33:696, 1976.
- Carter BS, Haverkamp AD, Merenstein GB. The definition of acute perinatal asphyxia. Clin Perinatol 20:287, 1993.
- Robertson CMT, Finer NN. Long-term followup of term neonates with perinatal asphyxia. Clin Perinatol 20:483, 1993.
- De Souza SW, Richards B. Neurologic sequelae in newborn babies after perinatal asphyxia. Arch Dis Child 53:464, 1978.
- Finer NN, Robertson CM, Richards RT et al. Hypoxic-ischemic encephalopathy in term neonates: Perinatal factors and outcome. J Pediatr 98:112, 1981.
- American Academy of Pediatrics/American College of Obstetricians and Gynecologists. Guidelines for Perinatal Care, 3rd edition. AAP/ACOG, 1992.
- Cohn EH, Sacks EJ, Jeyman MA et al. Cardiovascular response to hypoxemia and acidemia in fetal lambs. Am J Obstet Gynecol 120:817, 1974.
- Rudolph AM. The fetal circulation and its response to stress. J Dev Physiol 6:11, 1984.
- Portman RJ, Carter BS, Gaylord MS et al. Predicting neonatal morbidity after perinatal asphyxia: A scoring system. Am J Obstet Gynecol 162:174, 1990.
- APS/SPR Program. Pediatr Res 33 (Part 2):1A-432A, 1993.
- Perlman JM, Tack EM, Martin T et al. Acute system organ injury in term infants after asphyxia. Am J Dis Child 143:617, 1989.
- Warburton D, Singer DB, Oh W. Effects of acidosis on the activity of creatine phosphokinase and its isoenzymes in the serum of newborns. Pediatr 68:195, 1981.
- Fernandez F, Verdu A, Quero J et al. Cerebrospinal fluid lactate levels in term infants with perinatal asphyxia. Pediatr Neurol 2:39, 1986.
- Low JA. Lactate measurements in perinatology. In: Laursen NH, Hockberg HM (eds). Clinical Perinatal Biochemical Monitoring. Baltimore: Williams and Wilkins, 1981.
- Mathew OP, Bland H, Boxerman SB et al. CSF lactate levels in high-risk neonates with and without asphyxia. Pediatr 66:224, 1980.
- Suidan JS, Young BK. Outcome of fetuses with lactic acidemia. Am J Obstet Gynecol 150:33, 1984.
- McGuinness GA, Weisz SC, Bell WE. CSF lactate levels in neonates: Effect of asphyxia, gestational age, and postnatal age. Am J Dis Child 137:48, 1983.
- Thorpe JA, Sampson JE, Parisi VM et al. Routine umbilical cord blood gas determinations? Am J Obstet Gynecol 161:600, 1989.
- Yoemans ER, Hauth JC, Gilstrap LC III et al. Umbilical cord pH, PCO2 and bicarbonate following uncomplicated term vaginal deliveries. Am J Obstet Gynecol 141:798, 1985.
- Ruth VJ, Raivia KO. Perinatal brain damage: Predictive value of metabolic acidosis and the Apgar score. Br Med J 297:24, 1988.
- Goodwin TM, Belai I, Hernandez P et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol 162:1506, 1992.
- Winkler CL, Hauth JC, Tucker M et al. Neonatal complications at term as related to the degree of umbilical arterial acidemia. Am J Obstet Gynecol 164:637, 1991.
- Gilstrap LC III, Leveno KL, Burris J et al. Diagnosis of birth asphyxia on the basis of fetal pH, Apgar score and newborn cerebral dysfunction. Am J Obstet Gynecol 161:825, 1989.
- Niswander KR. EFM and brain damage in term and post-term infants. Contemp Obstet Gynecol 36:39, 1991.
- Quilligan EJ, Paul RH. Fetal monitoring, is it worth it? Am J Obstet Gynecol 45:96, 1975.
- Haverkamp AD, Thompson ME, McFee Jr et al. The evaluation of continuous fetal heart rate monitoring in high-risk pregnancy Am J Obstet Gynecol 125: 310, 1976.
- Shy KK, Larson EB, Luthy DA. Evaluating a new technology: The effectiveness of electronic fetal monitoring. Ann Rev Public Health 8:165, 1987.
- Freeman R. Intrapartum fetal monitoring—a disappointing story. N Engl J Med 322:624, 1990.
- Apgar V. A proposal for a new evaluation of the newborn infant. Curr Res Anesth Analg 32:260, 1953.
- Apgar V, Holiday DA, James LS et al. Evaluation of the newborn infant—second report. JAMA 168:1985, 1968.
- Butterfield LJ, Covey M. Practical epigram of Apgar score. JAMA 181:353, 1972.
- Paneth N, Fox HE. The relationship of Apgar score to neurologic handicap; a survey of clinicians. Obstet Gynecol 61:547, 1983.
- American Academy of Pediatrics, Committee on Fetus and Newborn. Use and abuse of the Apgar score. Pediatrics 78:1148, 1986.
- American Hospital Association. Apgar score use and issues. Coding Clinic for ICD-9-CM, 3:3, 1986.
- Lauerner PA, Calame A, Janacek P et al. Systemic pH measurements in the umbilical artery: Causes and predictive value of neonatal acidosis. J Perinat Med 11:278, 1983.
- Page FO, Martin JN, Palmer SM et al. Correlation of neonatal acid-base status with Apgar score and fetal heart rate tracings. Am J Obstet Gynecol 145:1306, 1986.
Discussion
John Freeman, M.D.
Johns Hopkins Hospital, Baltimore, Maryland
The purpose of this conference is to develop a research definition of perinatal asphyxia. Now why on earth would you want to do that? What is a research definition of asphyxia as opposed to a real definition; a legal definition; an obstetrical definition; or what ever.
We already know how to define asphyxia. The definition is in Webster's dictionary and in Dorland's. According to Webster's the word is derived from the Greek: asphyxia—stopping of the pulse, (a-sphyzein to beat violently) loss of consciousness as a result of too little oxygen and too much carbon dioxide in the blood: suffocation causes asphyxia. The problems seems to be the one articulated by Humpty-Dumpty to Alice when he said, "When I use a word it means just what I choose it to mean—neither more nor less." Our problem is not with what the words mean, but in choosing what we want them to mean.
Definitions will and should be partially dependent on the purpose for which we want to use them, and on the precision which we desire in their use. The literature and recent research has, unfortunately, used the term asphyxia in many different ways which vary from study to study, and which are usually used imprecisely, since there are no direct measures of asphyxia. But this imprecision has always been true of the clinical and research studies of neonatal asphyxia, just as it is for many other clinical conditions. Why have we suddenly discovered a need for a new research definition? One reason is that since the legal system has become interested in asphyxia, the medical profession has become enmeshed in the questions of when and if it occurred, and in its causes and consequences. Thus there are now legal consequences to any such definition.
We seem to want asphyxia to mean different things for different purposes. For legal purposes we would like asphyxia defined as— sufficiently severe lack of oxygen or of blood flow to result in brain damage with permanent motor impairment, often intellectual deficit and/or seizures. This neurologically oriented, legal definition would ignore the renal, cardiac and pulmonary dysfunctions which may accompany severe asphyxia, since these rarely are the cause of law suits. Such a definition, of severe, profound, or prolonged asphyxia, however, incorporates knowledge of the child's future, and/or an ability to look back at the perinatal period. It requires a dimension of time to know that the neurological damage was permanent and also implicitly embodies the ability to have ruled out other conditions which may mimic asphyxia. The dimension of time imposes constraints on the use of such a definition in incerventional research on the newborn, where time may be critical. It should also be noted that this definition of asphyxia does not embody concepts of either cause or fault.
Why do we need a research definition of asphyxia? We know how to produce asphyxia experimentally by depriving a cell system or an animal of oxygen (or blood supply), and studying what happens. We can also perturb what happens—or prevent what happens—by intervening with the administration of X, Y, or Q. What we can't do with cells or animals is to see if we are able to produce or prevent the one clear outcome of birth asphyxia that is of interest in human infants—cerebral palsy. To do that would require the use of primates, as was done by Windell and Myers, and primates are too difficult to work with in an ethical fashion, and too expensive. Therefore we have to turn to humans.
The FDA requires that before drugs may be used for humans, they must be tested in consenting adults, first for safety and then for efficacy Safety cannot be tested in healthy newborns since consent cannot be given. Studying efficacy first in this group requires a condition or disease which does not occur in adults.
Perhaps one could obtain permission to study interventions in the newborn with medications not previously tested in adults if the drug was used to prevent the consequences of an injury not seen in older children or adults. Is this true for hypoxic-ischemic injury? Medication might also be used if the child had clearly suffered sufficiently severe hypoxic-ischemic injury to be at very high risk developing permanent motor neurologic damage. That situation must be sufficiently clear that the risks and benefits of the intervention clearly outweigh those of alternative courses of action. Furthermore, the intervention must be performed sufficiently soon after the insult to have a high probability of preventing whatever it is that results in the permanent injury.
The medication in question would nevertheless have to be first tested for safety in consenting adults, (despite the fact that this would tell us little about its effect on the developing nervous system). It should also probably be studied for evidence of efficacy in semi-analogous situations such as drowning or strokes.
Our task, therefore, is not to develop a research definition of asphyxia, but to develop a means for the early identification of an infant who has suffered sufficiently severe asphyxia to have a substantial (how high) likelihood of suffering severe, permanent neurological deficit. At the same time we must be reasonably certain that the infant does not have another condition whose signs and symptoms mimic asphyxia. These criteria must be met soon (how soon?) after the asphyxial event, at a time when interventions could favorably alter outcome.
What then in the clinical assessment of the neonate permits such an early diagnosis, and is of sufficient precision to exclude most other conditions simulating or confused with asphyxia? Let us briefly discuss the three papers in this session and see how, or if they meet these criteria.
Merenstein and Carter aptly criticize the old ICD-9 coding. They present the new code, which is useless for the early diagnosis of asphyxia, and discuss the fact that "... a newborn with sufficiently severe hypoxia to result in HIE will show other signs of hypoxic damage including multiorgan system dysfunction." They then discuss the limitations of the infant's acid-base status, of the Apgar scores, and of electronic fetal monitoring. As they discuss the definition of asphyxia from the new Guidelines for Perinatal Care, they point out that observation for a variable time period is required. Once again, this definition is useful for some purposes, but not for others. It would be fine if the question was, "Could asphyxia have caused this child's neurologic deficit?" However, if the question is, "Has this infant incurred sufficient hypoxic-ischemic insult that there is a high probability that permanent neurologic damage will occur?", then the AAP/ACOG guidelines definition requires too much time. The scoring system proposed using: 1) the 5-minute Apgar score, looking at evidence of neurologic depression (why not use a 10-minute score?); 2) the base deficit, as evidence of prior oxygen/circulation dysfunction; and 3) the fetal heart tracing, as evidence of intrauterine fetal distress, possibly due to intrauterine circulatory compromise, would (or should) greatly enrich the population labeled asphyxia. Will it be sufficiently precise, specific, and useful to meet the tests discussed previously? Clearly it needs to be evaluated for its correlation with neonatal morbidity (neurologic as well as multi-organ), and long-term outcome.
Clancy carefully points out that the EEG is too sensitive to indicate the severity of the insult and too non-specific to, of itself, indicate etiology. This is a criticism which should be applied to many of the single landmarks (i.e., Apgar score, pH, time of resuscitation, etc.) that we have been utilizing in the past to define asphyxia. The changes in the EEG with time can give some indication of the trends in the brain's dysfunction, but even using the most abnormal EEG, even if it correlates well with outcome, will do little to advance our research definition of asphyxia or its early diagnosis. Worst requires repeated studies over time for assessment.
Nelson's paper leaves me perplexed. First, because he does not appear to be talking about the global fetal hypoxia which we are usually discussing, but rather about ischemia, and focal ischemia at that, a condition which is usually ignored in our discussions of HIE. In doing so he brings out a crucial element which we have omitted as we develop this research definition of asphyxia, the element of stroke in the perinatal period. The correlation of this entity to intrauterine problems has been poorly studied, and its etiology is largely unknown. However, if we are to intervene with medications, shouldn't we be evaluating the effects of our interventions on the hypoxic-ischemic neural tissue resulting from the stroke? Indeed, if that were one of our goals (as it should be) how does one diagnose stroke in the newborn? At what perinatal epoch does it occur? How much time is there after the event before the damage is permanent? Which interventions will improve outcome? These questions about the perinatal period are clearly just as applicable to infants, older children and adults with stroke, and are more easily studied in those populations. Perhaps those are the places we should begin to look at the effects of our interventions since we could avoid the constraints on neonatal research with new drugs.
Summary
Asphyxia is not an all-or-none phenomenon. It is a graded condition, starting with the hypoxic environment of the uterus. The clinical signs of its global effects are also graded: first neurologic and then, with increasing severity and duration of the hypoxia and ischemia, are systemic. The choice of when to intervene on this slippery slope depends on the certainty of the diagnosis which is desired; on the certainty and severity of the prognosis which you hope to prevent; and on the risks which are incurred by the intervention. None of these have to do with the development of a research definition of asphyxia.
Discussion
William Oh, M.D.
Women & Infants Hospital of Rhode Island, Providence
The goal of clinical assessment of an infant with perinatal depression at birth is twofold: 1) to establish the diagnosis of perinatal asphyxia, and 2) to gather information that will form the basis for parental counseling in regard to prognosis. The clinician's task in achieving such goal is often difficult for a number of reasons. The degree of fetal insults and the response to such insult varies and is highly unpredictable. The clinical and diagnostic data necessary to arrive at a diagnosis and allow the clinicians to make a reasonable prognostic estimate are often not available. Furthermore, even if data are available, the interpretation of this information is often fraught with the lack of a research basis for a reasonable conclusion.
With the aforementioned disclaimer, a clinician can still achieve the goal by collecting and making individual clinical judgment in assessing an infant with perinatal depression. The following table lists some of the pertinent data that may be useful in providing a comprehensive assessment of an infant under consideration for a diagnosis of perinatal asphyxia. This information can be used to provide a baseline in the continuing and prospective evaluation of the infant that will eventually allow for the establishment of prognosis and perhaps institute early intervention to improve outcome.
Using the clinical parameters and diagnostic methodologies listed in Table 1, a clinician can make a diagnosis of perinatal asphyxia in most cases. For instance, a full term infant born to a mother who has toxemia of pregnancy with a non reactive non-stress test demonstrates cord compression fetal heart rate pattern, thick meconium and a scalp pH of 7.10 just prior to delivery is likely to have fetal distress. If this infant has an Apgar score of 1 or 2 at 1 minute and requires aggressive resuscitation followed by an Apgar score of less than 4 at 5 minutes, the diagnosis of perinatal asphyxia would be very likely. If the postnatal course is complicated by significant seizure with abnormal EEG and there are signs of other organ injury (oliguria, feeding intolerance, respiratory distress with persistent pulmonary hypertension, etc.), the diagnosis of perinatal asphyxia can be quite definite.
In many cases, due to lack of adequate information, the diagnosis can be very difficult to make. This is in part due to variability in clinical events and inappropriate use of diagnostic tests both in the selection and the timing of performing these diagnostic methodologies. The discussion by the previous speakers are valuable with reference to the appropriate use and interpretations of cranial ultrasound, EEG, cranial CT scan and MRI.
A more difficult task with reference to perinatal asphyxia is the ability of the clinician to provide a prognostic statement based on clinical assessment during the perinatal period. In extreme cases (mildest and severest forms of perinatal asphyxia), a reasonable prognosis may be rendered; however in most cases, one often needs to provide a provisional prognostic statement (if at all possible) to the parents and reassess the situation with close follow-up and assessment. There is a great need for the development of research to explore the utility of non-invasive technologies that could help the clinicians in the assessment of these infants for diagnosis and prognosis.
TABLE 1: Diagnosis of Perinatal Asphyxia
| PERIOD | PARAMETERS | Clinical Diagnostic |
|---|---|---|
| Prenatal | Pregnancy complications, e.g., toxemia, PIH | Non-stress test, contraction test, biophysical profiles |
| Intrapartum | Meconium stain, abnormal labor, prolapsed cord, vaginal bleeding | Fetal heart rate pattern, scalp pH |
| Birth | Apgar scores, resuscitation requirements | Cord blood pH & base excess, cord blood chemistries, e.g., creatinine phosphate kinase |
| Postnatal | Organ related clinical morbidities: CNS, Renal, GI, etc. | Metabolic profiles, glucose, calcium, etc. Liver enzymes, EEG cranial ultrasound, CT scan, MRI. |
Session lV: Interventions
Moderator: Deborah Hirtz
Acute Perinatal Asphyxia—Conventional Management
James A. Lemons, M.D.
Section of Neonatal-Perinatal Medicine,
Indiana University Medical Center, Indianapolis
Introduction
Although the major components of conventional management of acute perinatal asphyxia in the term infant can be identified, there is little consensus regarding the best approach to the infant with severe birth depression. This is reflected clearly in a survey of 72 training programs in neonatal-perinatal medicine undertaken in 1988 by Donn et al.1 Of all the potential therapies identified, only fluid restriction and treatment of seizures with phenobarbital were considered standard among the institutions polled.
The diversity in practice reflects our lack of firm knowledge regarding the diagnosis, management, pathophysiology and long-term morbidity of perinatal asphyxia. Furthermore, effectiveness of care for the infant after birth is necessarily limited by the severity of the insult which has already occurred. Regardless of how good the postnatal interventions are, it is unlikely that they will be able to reverse or prevent permanent neurologic damage in many cases of severe asphyxic injury.
As thoroughly reviewed by Peliowski and Finer,2 even the most widely accepted interventions (e.g., fluid restriction) provided to infants with birth asphyxia lack appropriate evaluation through randomized trials. Most reported studies have been performed using relatively small sample sizes, an imprecise definition of asphyxia and without specific parameters to classify infants according to severity of insult. Study populations are therefore heterogeneous as investigators have been unable to document with certainty the severity of the insult, the duration of the insult, and/or the timing of the insult in relation to birth. In some instances other potential etiologies for the abnormalities noted in the neonatal period (e.g., depressed Apgar scores, acidosis, neurologic abnormalities) have not been excluded (i.e., anomalies, infection, metabolic disorders, drug ingestions, etc.)
As discussed elsewhere in this conference, scoring systems have been developed and tested which can aid in the quantitation of birth asphyxia for prospective study.3 The most accurate predictors of post-asphyxia morbidity, for example, appear to be a combination of the 5- minute Apgar, the base deficit measured within the first hour of life, and the fetal heart rate tracing abnormalities prior to birth.4 Long term outcome appears to be most closely related to the presence and severity of hypoxicischemic encephalopathy (HIE) manifest in the first 7 days of life as defined by Sarnat and Sarnat5 and as confirmed by Finer.6
Current Conventional Therapies
Obstetric Interventions
Prevention or interruption of an insult to the fetus has been discussed in other sections and may represent the most effective way to reduce the incidence of perinatal asphyxia and resultant HIE. In spite of comprehensive obstetric screening and monitoring, however, it may be expected that approximately 50 percent of cases of perinatal asphyxia may be unanticipated and unpreventable.7 Nonetheless, strategies to improve our monitoring capability of the fetus are of highest priority and are likely to be our most effective strategy toward prevention.
Care of the Newborn at Birth
Initial treatment of the asphyxiated newborn at birth is directed first at eliminating hypoxia/ hypercarbia by restoring appropriate oxygenation and ventilation, and second at preventing and alleviating tissue ischemia by insuring optimal tissue perfusion. Additional therapies are generally directed at specific complications or morbidities as they arise, such as seizures. If a particular organ is affected, as may occur with meconium aspiration or acute renal tubular necrosis, then treatment directed at those specific problems should be prioritized. Most other therapies intended to reduce acute and long term CNS morbidity remain controversial, some of which will be discussed in the following sections.
Neonatal Resuscitation
As previously mentioned, a need for resuscitation in the term pregnancy complicated by acute perinatal asphyxia may be anticipated in only 50 percent of the cases. Therefore, it is imperative that skilled personnel and all necessary equipment be available at every delivery to insure appropriate resuscitation of the infant if needed. As perinatal asphyxia occurs relatively infrequently (three to five per 1,000 live births), maintaining a high level of expertise for such resuscitation is often a challenge for many hospital staffs, particularly those with relatively small obstetric services.
Ventilation
In the vast majority of cases, the institution of assisted ventilation is all that is required for successful resuscitation. Establishment of effective ventilation requires clearing of excess fluid, or other debris, from the airway followed by application of appropriate positive pressure to open the lungs. Controlled trials have not been performed to document the most effective method of providing such assisted ventilation. However, it is clear that intubation is not necessary in the majority of cases and that appropriate ventilation may be achieved with an ambu bag, appropriately fitting mask and 100 percent oxygen.8 In infants who do not respond promptly to ambu, mask and oxygen, endotracheal intubation may be required. Again appropriate pressures must he applied and titrated to the individual infant as evidenced by movement of the chest and clinical response. The development of adequate functional residual capacity (FRC) appears to be important in establishing spontaneous respirations,9 and potentially in enhancing surfactant production.10 The procedures as recommended by the American Academy of Pediatrics/American Heart Association provide appropriate technique for effective resuscitation.11
Treatment of Metabolic Acidosis
The use of sodium bicarbonate remains controversial even in severe birth asphyxia. The primary potential benefits of sodium bicarbonate therapy in the presence of severe metabolic acidosis would include improved myocardial performance with secondary improvement in perfusion of vital organs, and theoretically a reduction in tissue ischemia with the possibility of improved long-term outcome. Hazards of bicarbonate infusion include a sudden increase in serum osmolality with risk of hemorrhage (particularly in the brain), sudden increase in venous pressure with reduction in CSF pressure, reduction in cerebral blood flow, and a transient increase in PCO 2, (dependent upon the ventilatory status of
the infant).12-14 It is apparent that with effective cardiopulmonary resuscitation, the metabolic and respiratory acidoses which accompany birth asphyxia will generally resolve over 20-40 minutes. However, the washout phenomena secondary to improved perfusion and transiently increased lactic acid levels may lead to a worsening in the acidosis temporarily. This should correct spontaneously if oxygenation and ventilation are adequate and cardiac and circulatory support are maintained. Although bicarbonate has not been proven to be of benefit in neonatal resuscitation,15,16 most clinicians will employ sodium bicarbonate as a slow infusion over at least 5-10 minutes in the presence of documented severe metabolic acidosis.
Circulatory Support
In the presence of cardiac arrest or severe bradycardia unresponsive to ventilation, cardiac massage should be instituted. In two reported studies, cardiac massage is most effective when the method of hands encircling the chest was used as compared to the two-finger technique of compression over the sternum.17 If appropriate ventilation/oxygenation and cardiac massage do not effect a prompt improvement in heart rate and blood pressure, then other interventions may be necessary. Intravenous or endotracheal epinephrine may result in prompt improvement in cardiac function.
In the presence of acute blood loss and shock, volume expansion is required. This can be achieved with a variety of agents, although in this instance whole blood is preferred. In the absence of evidence of shock and acute blood loss, the need for volume expansion may be determined on clinical grounds using blood pressure, peripheral perfusion, hematocrit and urine output as guides. Acutely, the assessment of blood gas status, blood pressure and peripheral perfusion will be most useful. In the presence of severe metabolic acidosis, there will often be peripheral vasoconstriction which may lessen as oxygenation and cardiac function improve. With a relaxation of the peripheral vascular bed, blood pressure may fall and perfusion may in fact worsen. In this instance, volume expansion may be appropriate, but should be undertaken cautiously. Hypervolemia may increase blood volume excessively and potentially result in high blood pressure. Recognizing that cerebrovascular autoregulation may be compromised by an asphyxic insult, systemic hypertension may increase the risk of intracerebral hemorrhage and other complications, such as patent ductus arteriosclerosis.18,19Nonetheless, acute support of vascular volume is often warranted during initial stabilization of the asphyxiated infant.
Use of other agents such as dopamine to support blood pressure and cardiac output may be appropriate. In infants who are asphyxiated and have compromised peripheral perfusion with hypotension, low dose dopamine has been demonstrated to increase systolic blood pressure.20 Potential benefits of dopomine include improved cardiac contractility as well as improved renal function in the early phase of recovery from the asphyxic insult.
Metabolic Support
An adequate supply of glucose is critical for the neonatal brain and the heart during the recovery from an asphyxial insult. However, it is also recognized that excessively high levels of glucose may have an adverse effect on the brain by inducing local lactic acidosis, in that the damaged areas of the brain may be unable to completely metabolize glucose. Generally it is recommended that adequate glucose infusion be initiated promptly in the asphyxiated infant to avoid hypoglycemia, while preventing hyperglycemia due to an excessive exogenous glucose load.
Benefit of calcium during early neonatal resuscitation has not been supported by clinical studies (although adequate controlled trials have not been undertaken). Hazards include bradycardia, tissue necrosis, and organ damage secondary to intravascular infusion.21 The AAP currently does not recommend calcium during newborn resuscitation.22
Other agents/strategies which have been advocated for resuscitation of the asphyxiated infant include naloxone, hyperbaric oxygen and hypothermia. None of these has been demonstrated to be safe or effective in randomized controlled trials, and therefore are not recommended at this time.23,24
Treatment of HIE
In the presence of moderate to severe birth asphyxia, primary attention is given to minimizing the severity of HIE which may develop over the ensuing week of life. A number of strategies have been proposed which may ameliorate the severity of encephalopathy and potentially improve long-term outcome.
Fluid restriction is generally recommended for all infants with moderate or severe perinatal asphyxia after initial resuscitation and stabilization. Because of the potential for renal injury with oliguria, as well as for inappropriate secretion of antidiuretic hormone, fluid intake is normally restricted to meet estimated rates of insensible water loss. Such fluid restriction is intended to minimize complications of hypervolemia, which may include cerebral edema. Although controlled trials have not been reported which document the efficacy of this measure, common sense would indicate that this is a rational approach to infants with more severe degrees of birth depression. Restriction of fluid must be undertaken carefully, insuring that adequate blood volume, blood pressure, and tissue perfusion are maintained. A hematocrit in the normal range is also of importance.
Perinatal asphyxia accounts for the majority of neonatal seizures, which occur in 50-70 percent of infants with moderate to severe perinatal insult.25 In most cases, seizures caused by asphyxia will develop between 12-24 hours of age, but may occur earlier in the more severe cases, depending upon the time of the insult.26 At this time, no studies in human infants have confirmed that seizures in and of themselves aggravate the injury to the brain—even if they are prolonged or repetitive. However, several animal studies suggest that status epilepticus may lead to long-lasting alterations in brain development, implying that seizures may increase the risk of long-term CNS sequelae.27,28 Most experts agree that in spite of the lack of objective evidence, vigorous treatment of seizures in the newborn period with appropriate anticonvulsants is warranted. Barbiturates are generally accepted as the agent of choice at this time. Barbiturates decrease the metabolic rate and energy utilization of neuronal and glial tissue in a variety of ways, reducing the need of the brain for glucose and oxygen.29,30 Other demonstrated effects of phenobarbital include decreased cerebral blood flow, (possibly mediated by reduced metabolic rate of the neural tissue and, therefore, reduced demand for energy substrate), and possible scavenging of toxic-free radicals. Each of these effects theoretically may benefit the acutely injured brain and potentially protect the central nervous system from further damage at a time when cerebral perfusion and substrate delivery may be compromised by edema, hypotension, and loss of autoregulation. Vigorous treatment of seizures may require multiple doses of phenobarbital, and serum levels which exceed 60 m cg/ml. Even these high levels may not effectively eliminate seizures, but often will reduce the frequency of convulsions during the acute post-asphyxial period. High phenobarbital concentrations may also lead to sedation of the infant and possibly hypotension through myocardial suppression. Careful attention must be paid to maintaining appropriate ventilation and oxygenation, as well as ensuring support of the cardiovascular system during treatment. Often phenobarbital may only be required for treatment of postasphyxial seizures during the first several days of life. Normally the seizures abate after 48-72 hours and are not likely to reoccur.
Prophylactic treatment of infants with barbiturates following severe asphyxia but prior to onset of seizures has not been shown to alter outcome.31 Furthermore, complications of hypocension are significantly more common in the infants treated with barbiturate. Therefore, prophylactic treatment with phenobarbital or other barbiturate is not recommended at this time.
Other strategies to reduce cerebral injury secondary to edema have been proposed. These include hyperventilation, steroids, osmotic agents and diuretics. None of these, with the exception of mannitol, have been found to be effective and are therefore not recommended for routine use in the newborn for treatment of asphyxia. Studies of mannitol used in small numbers of infants suggest that intracranial pressure may be effectively decreased with this agent.32 Although these reports are provocative, the studies to date have failed to carefully define the infant populations by degree of asphyxia, and therefore, the results cannot be extrapolated to support routine use of mannitol at this time. These data would suggest that larger randomized, controlled trials may be appropriate.
Conclusion
Conventional management of the term infant with perinatal asphyxia appears to be rather straightforward. Prompt and expert resuscitation, meticulous attention to support of the cardiovascular and respiratory systems, careful monitoring of basic metabolic and hematopoietic parameters, vigorous treatment of seizures with appropriate medications (primary agent being phenobarbital), appropriate attention to other organ system injuries (particularly renal, cardiac and intestinal), and avoidance of unproven therapies will optimize outcome. Excellent care relies upon timely intervention with good tchnique, experience and common sense. Newer therapies warrant careful investigation as the mechanism underlying the cellular and molecular pathophysiology of asphyxia become more clearly delineated. Ultimately prevention of the injury may be the most effective, yet the most elusive, strategy to decrease perinatal morbidity secondary to birth asphyxia.
References
- Donn SM, Goldstein GW, Schork MA. Neonatal hypoxic-ischemic encephalopathy: Current management practices. J Perinatol 8:49-52, 1988.
- Peliowski A, Finer NN. Birth asphyxia in the term infant. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, pp. 249-279, 1992.
- Carter BS, Haverkamp AB, Merenstein, GB. The definition of acute perinatal asphyxia. Clin Perinatol 20:287-304, 1993.
- Portman RJ, Carter BS, Gaylord MS et al. Predicting neonatal morbidity after perinatal asphyxia: A scoring system. Am J Obstet Gynecol 162:174-182, 1990.
- Sarnat HB, Sarnat MS. Neonatal encephalopathy following fetal distress. Arch Neurol 33:696, 1976. In: Shankaran S (ed). Clinics in Perinacology: Perinatal Asphyxia. Philadelphia: W.B. Saunders, p 303, 1993.
- Finer NN, Robertson CM, Richards RT et al. Hypoxemic-ischemic encephalopathy in term neonates: Perinatal factors and outcome. J Pediatr 98:112, 1981. In: Shankaran S (ed). Clinics in Perinatology: Perinatal Asphyxia. Philadelphia: W.B. Saunders, p 303, 1993.
- Jacobs MM, Phibbs RH. Prevention, recognition, and treatment of perinatal asphyxia. Clin Perinatol 16:785-807, 1989.
- Milner A, Vyas H, Hopkin I. Efficacy of face mask resuscitation at birth. Br Med J 289:1563-1565, 1984. In: Sinclair JC, Bracken, MB. Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 274, 1992.
- Boon A, Milner A, Hopkin I. Physiological responses of the newborn infant to resuscitation. Arch Dis Child 54:492-498, 1979.
- Massaro GD, Masaro D. Morphologic evidence that large inflations of the lung stimulate secretion of surfactant. Ann Rev Respir Dis 127:235-236, 1983. In: Clinics in Perinatology, Critical Issues in Intrapartum and Delivery Room Management. Philadelphia: W.B. Saunders, p 805, 1989.
- Bloom RS, Cropley C (eds). Textbook of Neonatal Resuscitation. American Heart Association/American Academy of Pediatrics. Dallas, p 275, 1987. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 275, 1992.
- Kravath RE, Aharon AS, Abal G, Finberg L. Clinically significant physiologic changes from rapidly administered hypertonic solutions: Acute osmol poisoning. Pediatrics 46:267-275, 1970. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Simmons MA, Adcock EW, Bard H, Battaglia FC. Hypernatremia and intracranial hemorrhage in neonates. New Engl J Med, 291:6-10, 1974. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Lou HC, Lassen NA, Friis-Hansen B. Decreased cerebral blood flow after administration of sodium bircarbonate in the distressed newborn infant. Acta Neurol Scand 1978; 57:239-247. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Corbet AJ, Adams JM, Kenny JD et al. Controlled trial of bicarbonate therapy in high-risk premature newborn infants. J Pediatr 91:771-776, 1977. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Bland RD, Clarke T, Harden LB. Rapid infusion of sodium bicarbonate and albumin into high-risk premature infants soon after birth: A controlled, prospective trial. Am J Obstet Gynecol 124:263-267, 1976. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- David R. Closed chest cardiac massage in the newborn infant. Pediatrics 81:552-554, 1988. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 275, 1992.
- Goldberg RN, Chung D, Goldman S, Bancalari E. The association of rapid volume expansion and intraventricular hemorrhage in the preterm infant. J Pediatr 96:1060-1063, 1980. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Godard-Finegold J, Armstrong D, Zeller RS. Intraventricular hemorrhage following volume expansion after hypovolemic hypotension in the newborn beagle. J Pediatr 100:796-799, 1982. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- DiSessa TG, Leitner M, Ti CC et al. The cardiovascular effects of dopamine in the severely asphyxiated neonate. J Pediatr 99:772-776, 1981. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Book LS, Herbst JJ, Stewart D. Hazards of calcium gluconate in the newborn infant: Intraarterial injection producing intestinal necrosis in rabbit ileum. J Pediatr 92:793-797, 1978. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- American Academy of Pediatrics, American College of Obstetricians and Gynecology: Guidelines for Perinatal Care. Evanston, Illinois, 1988. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 35, 1992.
- Chernick V, Manfreda J, DeBooy V et al. Clinical trial of naloxone in birth asphyxia. J Pediatr 223:519-525, 1988. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 37, 1992.
- Hutchison J, Kerr M, Inall J, Shanks R. Controlled trials of hyperbaric oxygen and tracheal incubation in asphyxia neonatorum. Lancet 1:935-939, 1966. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 275, 1992.
- Volpe JJ. Neurology of the Newborn, 2nd edition. Philadelphia: W.B. Saunders Company, p 236-279, 1987.
- Volpe JJ. Neonatal seizures: Current concepts and revised classification. Pediatrics 84:422-428, 1989.
- Wasterlain CG. Effects of neonatal status epilepticus on rat brain development. Neurol 26:975-986, 1976.
- Wasterlain CG. Effects of neonatal seizures on ontogeny of reflexes and behavior. Eur Neurol 15:9-19, 1977.
- Steen PA, Michenfelder JD. Mechanisms of barbiturate protection. Anesthesiol 53:183-185, 1980. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 276, 1992.
- Shapiro HM. Barbiturates in brain ischemia. Br J Anaesth 57:82-95, 1985. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 276, 1992.
- Goldberg R, Moscoso P, Bauer C et al. Use of barbiturate therapy in severe perinatal asphyxia: A randomized controlled trial. J Pediatr 109:851-856, 1986. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 276, 1992.
- Levene M, Evans D. Medical management of raised intracranial pressure after severe birth asphyxia. Arch Dis Child 60:12-16, 1985. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, p 276, 1992.
Neuronal Rescue and Neuronal Prophylaxis—Studies in Animals
Peter D. Gluckman, M.D., Christopher
E. Williams, Ph.D., Barbara M. Johnston,
D.Phil., Jian Guan, M.B., Ernest S. Sirimanne,
M.Phil., William K.M. Tan, Ph.D.
Research Centre for Developmental Medicine
and Biology, University of Auckland, New Zealand
Introduction
As reviewed elsewhere in this meeting, there are several primary considerations in addressing neuroprotective or neuronal rescue strategies. The most important is the relationship of the time of intervention to the putative insult. Different mechanisms dominate in neuronal injury during the insult itself as to those operative in the phase of delayed neuronal death. Thus IGF-1 which presumably interferes with the apoptotic cascade is effective if given after but not before an injury. Unfortunately this consideration has been frequently ignored leading to difficulty of interpretation of many studies and making it impossible to extrapolate to potential clinical application.
Secondly the confounding effect of sensitizing factors may influence the choice of therapy and interpretation of experimental data. For example, a calcium channel blocker such as flunarizine or nimodipine may be neuroprotective,1,2 but the secondary effects on the cardiovascular system may lead to hypotension and aggravation of the insult.3,4 Some agents used experimentally have caused hypothermia and it has been the hypothermia rather than the primary effect of the agent on the neuron that has led to apparent neuroprotection. In one of our studies we observed with MK-801 an increased mortality in the treated rather than control rats which led to a false impression of protection in the surviving treated rats. This review will focus on those studies which address those issues.
The mechanisms of neuronal death secondary to asphyxia are addressed elsewhere. Briefly the major mechanisms involved which might be addressed therapeutically include membrane instability, calcium toxicity excitotoxicity, free radical and other cycotoxin production, macrophage/microglial activation and apoptosis. In terms of primary neuronal loss it is likely that membrane instability and calcium toxicity offer the most interesting therapeutic possibilities for neuroprophylaxis. Inhibition of free radical production and or action is clearly of potential importance in the reperfusion phase. Seizure prophylaxis5 and inhibition of macrophage activation and the use of neurotrophic agents (presumably acting via inhibition of apoptosis)6 are likely to be the main basis of neuronal rescue therapies. We have previously reviewed some of the issues that must be considered in extrapolating from the experimental to the clinical situations.7,8
Interpretation of the Experimental Data
Despite the large number of studies in which potential neuronal protective effects are being addressed, many of the studies are of limited value. Relatively few consider the issue of timing of the intervention relative to the insult and often the relevance of output measures considered is limited. For example the effects of treatment on endpoints such as long-term functional recovery need to be determined. The question of the nature of the insult must be considered. Clearly there are differences in the mechanisms operative in the core of a focal insult to those in the penumbra or with more global insults9—the latter are probably those of most interest in the perinatal period—and it is essential to recognize whether there are systemic confounders (e.g., cardiovascular compromise) operative or not.10,11 The issue of developmental status is clearly important.12
Neuronal Prophylaxis
Strategies Aimed at Enhancing Membrane Stability
Two classes of agents have been described which probably act via increasing membrane stability: the gangliosides13 and the 21 amino steroids (lazeroids). The available data suggest both get incorporated into the membrane14 and are presumed to offer protection against membrane disruption associated with intracellular edema and lipid peroxidation by free radicals.13,15 GM 1 ganglioside confers considerable prophylactic protection in the fetal sheep subject to either prolonged cerebral ischemia16 or to repeated short episodes of ischemia. In the latter case the GM 1 ganglioside was infused after the first occlusion, mimicking a likely clinical paradigm of severe fetal distress, and conferred near-total protection.17 As GM 1 appeared to have no systemic side effects at least in the fetal sheep16 and is likely to cross the placenta18 this might offer an approach to emergency intervention prior to cesarean section. The data on lazeroids is more limited to short-term outcome but appears to have a similar mode of action.
Hypothermia
A number of experimental studies clearly show that mild hypothermia during an injury confers a protective advantage.
Glutamate Antagonists
Both NMDA receptor and kainic acid receptor antagonists have been shown to reduce neuronal loss when given before or after an hypoxic-ischemic injury.19-23 The efficacy depends on the nature of the injury and time of administration. For example the NMDA antagonists tend to be more effective when given before focal injuries rather than global ischemia.22 The NMDA receptor antagonists have been more widely studied in the immature brain before and after focal injuries. It would appear that some of these antagonists may have a therapeutic role but toxic or neurological side effects will probably limit their use.23-25
Calcium Channel Antagonists
Many studies have shown calcium channel blockers to be effective if given prior to an asphyxial injury.1,2,26However they are generally without effect if given after the injury,27 suggesting little role in delayed neuronal death. Further, at high dose, they may be cardiac depressants and aggravate the injury.26 We have observed a toxic interaction between flunarazine and MK801.28
Free Radical Scavengers and Synthesis Inhibition and Related Approaches
Free radical scavengers such as superoxide dismutase have a significant protective effect if given in the periasphyxial period.29,30 Their value in delayed neuronal death has not been elucidated. The use of allopurinol as an inhibitor of free radical production from purines has been suggested by one animal study31 and is being evaluated clinically. In the overall spectrum of the mechanisms of injury it is unlikely to have major effect. Similarly dexamethasone confers some protective effect in the immediate insult period.32
Neuronal Rescue
Immune Modulation
We have shown that TGFB1 is induced after hypoxic-ischemic injury and that exogenous TGFI3~ will reduce brain injury when given after the insult.33, 34 This is perhaps due to it suppressing microglial activation. Alternatively it might involve TGFb induction of the IGF system as is reported in fibroblasts. There is other experimental evidence suggesting that inhibition of macrophage activation is of therapeutic benefit.35
Anticonvulsants
Our studies in fetal sheep following cerebral ischemia clearly show some protective effect when MK801 is given 6 hours post-injury to suppress the seizures.5 Aggressive anticonvulsant therapy with EEG monitoring must be considered.
Neurotrophins
Our own studies have clearly shown that the IGF-1 system is induced in the region of neuronal loss after hypoxic-ischemic injury.6, 36 We have shown that IGF-1 given centrally after but not before the injury leads to histological6 and functional improvements (unpublished). The effect is mediated through the type I IGF receptor and explains earlier observations that insulin at high dose is neuroprotective. The effect probably depends on IGF-1 associating with the induced IGF binding proteins BP-2 and BP-3. Recently we have shown with tritiated IGF-1 that it preferentially concentrates in the region of injury (unpublished observations). Indirect evidence suggests that the action of IGF- 1 is to inhibit apoptosis. In contrast NGF is not neuroprotective in similar paradigms (unpublished data). There are reports of a neuronal protective effect of basic FGF37 and at least one study suggests this is dependent on the induction of IGF-1.38 This growth factor-dependent neuronal rescue appears to reflect endogenous neuroprotective mechanisms, suggesting it is a rational approach to neuronal rescue.
Apoptosis Inhibition
Growth factors and protein synthesis inhibitors can ameliorate cell death in vivo.39 Further there is evidence that IGF-1, which can inhibit apoptosis in vitro, can reduce neuronal injury when administered after the insult.6 Given that apoptosis is more likely to occur after moderate injuries,40,41 and necrosis is more likely to follow more severe injuries, factors that interfere with apoptosis may be more effective following the former.
Conclusions
Extrapolation from animal data to the clinical setting must be done with caution. There are many issues that must be considered when interpreting experimental data. The most important issues must be the risk of side effects or toxic interactions, particularly during situations such as global asphyxia or IUGR. Good clinical experimental design will require designing paradigms in which the temporal relationship of the insult and intervention is understood and the appropriate intervention is chosen together with appropriate outcome measures.
Acknowledgments
The authors work is supported by grants from the Health Research Council of New Zealand and the Neurological Foundation of New Zealand.
References
- Silverstein FS, Buchanan K, Hudson C, Johnston MV. Flunarizine limits hypoxia-ischemia induced morphologic injury in immature rat brain. Stroke 17:477-482, 1986.
- Gunn AJ, Mydlar T, Bennet L et al. The neuroprotective actions of a calcium channel antagonist, flunarizine, in the infant rat. Pediatr Res 25:573-576, 1989.
- Gunn AJ, Tan WKM, Gluckman PD, Williams CE. Neuroprotection of the fetus at risk of asphyxic injury. In: International Conference on Neuroprotective Agents, Rockland, Maine, 1991.
- Levene MI, Gibson NA, Fenton AC et al. The use of a calcium channel blocker, nicardipine, for severely asphyxiated newborn infants. Dev Med Child Neurol 32:567-574, 1990.
- Tan WKM, Williams CE, Gunn AJ et al. Suppression of post-ischemic epileptiform activity with MK-801 improves neural outcome in fetal sheep. Ann Neurol 32:677-682, 1992.
- Gluckman PD, Klempt ND, Guan J et al. A role for IGF-1 in the rescue of CNS neurons following hypoxic-ischemic injury. Biochem Biophys Res Commun 182:593-599, 1992.
- Gluckman PD, Williams CE. Is the cure worse than the disease? Caveats in the move from the laboratory to clinic. Dev Med Child Neurol 34:1015-1018, 1992.
- Gluckman PD, Williams CE. When and why do brain cells die? Dev Med Child Neurol 34:1010-1014, 1992.
- Choi DW. Cerebral hypoxia: Some new approaches and unanswered questions. J Neurosci 10:2493-2501, 1990.
- Soothill PW, Nicolaides KH, Campbell S. Prenatal asphyxia, hyperlactemia, hypoglycemia and erythroblastosis in growth retarded fetuses. BMJ 294:1051-1053, 1987.
- Thordstein M, Kjellmer I. Cerebral tolerance of hypoxia in growth-retarded and appropriately grown newborn guinea pigs. Pediatr Res 24:633-638, 1988.
- McDonald J, Johnston M. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Research Reviews 15:41-70, 1990.
- Mahadik SR Bharucha VA, Stadlin A et al. Loss and recovery of activities of a+ and a isozymes of (Na+ + K+)-ATPase in cortical focal ischemia: GM 1 ganglioside protects plasma membrane structure and function. J Neurosci Res 32:209-220, 1992.
- Toffano G, Benvegnu D, Bonetti AC et al. Interaction of GM 1 ganglioside with crude rat brain neuronal membranes. J Neurochem 35:861- 866,1980.
- Mahadik SP, Hawver DB, Hungund BL et al. GM 1 ganglioside treatment after global ischemia protects changes in membrane fatty acids and properties of Na+, KATPase and Mg2+-ATPase. J Neurosci Res 24:402-412, 1989.
- Tan WKM, Williams CE, Gunn AJ et al. Pretreatment with monosialoganglioside GM 1 protects the brain of fetal sheep against hypoxic- ischemic injury without causing systemic compromise. Pediatr Res 34:18-22, 1993.
- Tan WKM, Williams CE, Gunn AJ et al. Monosialoganglioside, GM 1 attenuates postischemic neuronal damage in the fetal sheep. Am J Obstet Gynecol 170:668-669, 1994.
- Magal E, Louis JC, Aguilera J, Yavin E. Gangliosides prevent ischemia-induced down-regulation of protein kinase C in fetal rat brain. J Neurochem 55:2126-2131, 1990.
- Sheardown MJ, Nielsen EO, Hansen AJ et al. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: A neuroprotectant for cerebral ischemia. Science 247:571-574, 1990.
- Buchan AM, Xue D, Huang ZG et al. Delayed AMPA receptor blockade reduces cerebral infarction induced by focal ischemia. Neuroreport 2:473 -476, 1991.
- Nellgard B, Wieloch T. Postischemic blockade of AMPA but not NMDA receptors mitigates neuronal damage in the rat brain following transient severe cerebral ischemia. J Cereb Blood Flow Metab 12:2-11, 1992.
- Buchan A, Li H, Pulsinelli WA. The N-methyl-D-aspartate antagonist, MK-801, fails to protect against neuronal damage caused by transient, severe forebrain ischemia in adult rats. J Neurosci 11:1049-1056, 1991.
- Gorter JA, Veerman M, Mirmiran M et al. Spectral analysis of the electroencephalogram in neonatal rats chronically treated with the NMDA antagonist MK-801. Dev Brain Res 64:37-41, 1991.
- Barth T, Grant ML, Schallert T. Effects of MK-801 on recovery from somatosensory cortex lesions. Stroke 21 (suppl III):153-157, 1990.
- Levene M. Role of excitatory amino acid antagonists in the management of birth asphyxia. Biol Neonate 62:248-251, 1992.
- Gunn AJ, Mallard EC, Williams CE, Gluckman PD. Effects of flunarizine therapy in cerebral ischemia in the fetal sheep. Pediatr Res 35:657 -660, 1994.
- Gunn AJ, Gluckman PD. Flunarizine, a calcium channel antagonist, is not neuroprotective when given after hypoxia-ischemia in the infant rat. Dev Pharmacol Ther 17:205-209, 1991.
- Gunn AJ, Gluckman PD et al. Unpublished observation.
- Imaizumi S, Woolworth V, Fishman RA et al. Liposome-entrapped superoxide dismutase reduces cerebral infarction in cerebral ischemia in rats. Stroke 21:1312-1317, 1990.
- Chan PH. Antioxidant-dependent amelioration of brain injury: Role of CuZn-superoxide dismutase. J Neurotrauma 9:S417-S423, 1992.
- Patt A, Harken A, Burton L et al. Xanthine oxidase-derived hydrogen peroxide contributes to ischemia reperfusion-induced edema in gerbil brains. J Clin Invest 81:1556-1562,1988.
- Tuor UI, Simone CS, Arellano R et al. Glucocorticoid prevention of neonatal hypoxic-ischemic damage: Role of hyperglycemia and antioxidant enzymes. Brain Res 604:165-172, 1993.
- McNeill H, Guan J, Miller O et al. Neuronal rescue with transforming growth factor beta 1 after hypoxic-ischemic brain injury. Neuroreport 5:901-904, 1994.
- Gluckman PD, Williams CE, McNeill H et al. Insulin-like growth factor 1 (IGF-1) and transforming growth factor beta 1 (TGFB1) rescue asphyxiated neurones when administered centrally. Endocrinol (abstr.), 1992.
- Giulian D, Robertson C. Inhibition of mononuclear phagocytes reduces ischemic injury in the spinal cord. Ann Neurol 27:33-42, 1990.
- Klempt ND, Klempt M, Gunn AJ et al. Expression of insulin-like growth factor binding protein-2(IGF-BP-2) following transient hypoxia- ischemia in the infant rat brain. Mol Brain Res 15:55-61, 1992.
- Yamada K, Kinoshita A, Kohmura E et al. Basic fibroblast growth factor prevents thalamic degeneration after cortical infarction. J Cereb Blood Flow Metab 11:472-478, 1991.
- Chernausek SD. Insulin-like growth factor-I (IGF-l) production by astroglial cells: Regulation and importance for epidermal growth factor-induced cell replication. J Neurosci Res 34:189-197, 1993.
- Shigeno T, Yamasaki Y, Kato G et al. Reduction of delayed neuronal death by inhibition of protein synthesis. Neurosci Lett 120:117-119, 1990.
- Okamoto M, Matsumoto M, Ohtsuki T et al. Apoptosis as an underlying mechanism of delayed neuronal death. J Cereb Blood Flow Metab 13:Abstract XI-12-S 80 (abstr.), 1993.
- Raff MC. Social controls on cell survival and cell death. Nature 356:397-400, 1992.
Glutamate Antagonists, Calcium Channel Blockers and Allopurinol
Malcolm I. Levene, M.D.
The General Infirmary at Leeds, England
Calcium and Neuronal Injury
Neuronal depolarization results in calcium entry into the neuron with secondary changes in cellular metabolism and in some cells restructuring of cytoarchitecture. This is thought to be the basis of both learning and neuronal plasticity. Calcium entry from the extracellular to the intracellular compartment occurs as the result of two distinct transport systems involving opening of ionic channels. These are voltage-sensitive (VSCC) and agonist-operated calcium channels (AOCC). VSCCs are present in both pre- and post-synaptic cerebral dendrites. At least three different types of VSCC within neurons have been recognized; T, N and L. Type N appears to be responsible for presynaptic release of neurotransmitter and type L on the postsynaptic dendrite mediates calcium entry. Type T has also been recognized in some CNS neurons. Stimulation of VSCC causes rapid entry of Ca2+ into the cell.
The major calcium port within central neurons is the AOCC which is stimulated by the excitatory amino acid neurotransmitter glutamate. Stimulation of postsynaptic receptors by glutamate activates two ionotropic receptors mediated by quisqualate/kainate (Q/K) and N-methyl-D- aspartate (NMDA). Glutamate stimulation initially opens Na+ and K+ ports causing cellular depolarization and causing fast excitation. Depolarization causes the Mg2+ gate on the NMDA port to open allowing Ca2+ to enter. The immature brain appears to have proportionately more NMDA receptors than the mature brain.
Effects of Asphyxia
Asphyxia causes excessive entry of calcium into the neuron. This appears to be mainly related to AOCC stimulation due to over-release of glutamate with consequent uncontrolled Ca2+ entry into the cell. This results in abnormal intracellular biochemical cascades with activation of proteases, lipases and endonucleases. ATP production is blocked, culminating in disruption of cytocellular architecture and cell death. A smaller component of Ca2+ entry appears to relate to enhanced porosity of VSCCs and associated failure of ATP production preventing calcium to be extruded from the cell.
Neuroprotection Directed Towards Ca2+ Entry
If intraneuronal Ca2+ entry is a major contributor to cell death then prevention of excessive accumulation may modify the fate of the neuron following global asphyxia. Much research in recent years has been focused on this area. Two main approaches directed towards both the AOCC and VSCC have been the subject of many reports.
NMDA Receptor Antagonists
A group of drugs have been shown to be NMDA antagonists and these can be divided into competitive and non-competitive types. 2-amino-5- phosphonoheptanoate directly blocks the glutamate site but is a relatively polar molecule and does not cross the blood-brain barrier. Soluble non-competitive NMDA antagonists have better penetration and these agents include ketamine, phencyclidine, dextromethorphan and dibenzocycloheptenimine known as MK-801.
There are a number of studies which suggest that NMDA receptor antagonists given before or after experimental hypoxic-ischemic insult (HII) protects the developing brain. The best studied antagonist is MK-801 which when given immediately after direct stereotactic injection of NMDA into a cerebral hemisphere in a 7-day old rat pup gave 95 percent protection against neuronal necrosis.1 Even when given 120 minutes after the insult there was 70 percent protection with the hippocampus.2 There appears to be a hierarchy of effect with MK-801 being the most effective, followed by CPP, PCP and TCP.3 CPP was found to be 14 times less potent than MK-801 in protecting against the direct NMDA lesion. Ketamine has been shown to have a neuroprotective effect when given 15 minutes after the NMDA brain insult1 but was far less effective than MK-801 and still somewhat less effective than CPP. MK-801 achieved 52 percent protection against cortical infaretion when given 1 hour after HII, but was ineffective at 4 hours post-insult.4
Unfortunately, MK-801 and other NMDA receptor antagonists are highly toxic. In one study of the protective effects of 10 mg/kg of MK-801, the mortality in 7-day old rat pups was 60 percent compared with only 13 percent in the control group.4 Others have shown that the mortality of MK-801 is dose-related and a dose of 1 mg/kg is associated with a lower mortality rate3 although even this dose is associated with a doubling in mortality compared with saline injected controls.5 In addition, MK-801. induces considerable neurobehavioural abnormalities in exposed rat pups including hyperexcitation initially followed by sedation and hypotonia.2
Mg2+ is a naturally occurring NMDA antagonist with a receptor site deep within the NMDA-dependent calcium channel. The maintenance of an elevated extracellular Mg2+ concentration protects the brain from experimental HII. Administration of magnesium sulphate up to 1 hour after exposure to excessive concentration of an NMDA-like compound protected the animal against neurologic sequelae.6 Magnesium sulphate has been used for over 60 years in perinatal medicine as a treatment for premature labour and severe preeclamptic toxaemia and seems to be well tolerated by the fetus and newborn although transient hypotonia and lethargy are commonly seen for a few days after exposure. We plan to start an international multicentre double-blind clinical controlled study evaluating the role of MgSO4 in severely asphyxiated newborn infants later this year.
Calcium Channel Blockers
As mentioned above Ca2+ entry through VSCCs may also be an important contributory cause of neuronal death following HII. A wide range of drugs have been assessed in this role under the general heading of calcium channel blockers. Unfortunately, the CCBs have a wide range of action on many cells and even within the nervous system the effect of CCBs vary depending on their relative affinity for different VSCC subtypes.
The affinity of CCBs on neuronal T-type channels has been studied by Akaike et al.7 and the order of blocking efficiency has shown to be flunarizine>nicardipine>nifedipine>nimodipine. T-type calcium channels have been suggested to be most closely associated with calcium-mediated neuronal toxicity8 and it is of interest that flunarizine and nicardipine appear to be the most potent inhibitors. There is some evidence that nicardipine also reduces the severity of cerebral oedema in a rat model of H II.9 A new generation of CCBs appear to have actions on both Na+ and Ca2+ ports with the effect of reducing depolarization and inhibiting secondary Ca2+ entry.9 An additional potentially protective action of the CCBs is an antioxidant effect.10
There have been a number of studies assessing the effects of a variety of CCBs in perinatal animals. Thiringer et al.11 showed that lidoflazine, when given together with oxygen-free radical scavengers, prevented post-asphyxial hypoperfusion in newborn lambs. Flunarizine, when given prior to HII, prevented post-asphyxial hypoperfusion in immature rat pups.12,13 In contrast to these studies, Ment et al.14 showed no improvement in cerebral blood flow in newborn beagle puppies following nimodopine exposure.
Only one study of the effects of a CCB in the human neonate has been published. Levene et al.15 gave the water soluble CCB, nicardipine, to four severely asphyxiated term neonates. Two of the four developed acute and severe hypotension with cardiovascular collapse following the start of the infusion. This is probably due to a negative inotropic effect on the heart as well as the vasodilating effect on vascular smooth muscle. Whether there was an additional effect on asphyxiated myocardium cannot be determined. Although hypotension is an anticipated but potentially dangerous effect of the CCBs, new drugs may became available with specific actions on neuronal T-type calcium channels.
Allopurinol
The production of free radicals is particularly likely to occur as the result of HII and are thought to play a major role in the pathophysiology of post-asphyxial damage. During hypoxia, ATP is degraded with the production of adenosine, inosine and hypoxanthine. With resuscitation and reperfusion the enzyme xanthine oxidase uses any available oxygen to convert hypoxanthine or xanthine to uric acid with the production of large quantities of oxygen free radicals. Free radicals are produced at many sites during the reoxygenation phase of an asphyxial injury. In the brain endothelial cells are the main source of xanthine oxidase and following asphyxia a xanthine oxidase inhibitor reduced the generation of free radicals from endothelial sites by 80-90 percent.16 There is some evidence that free radical generation can occur during HII and prior to reoxygenation,17 but in a neonatal animal model lipid peroxidation injury occurred only after an ischaemic insult.18
Free radical generation in the cerebral cortex and within the cerebrovascular endothelium may be particularly important in the development of irreversible cerebral injury. Allopurinol, a xanthine oxidase inhibitor, when given prior to the HII has been shown to substantially reduce neuronal necrosis, cerebral oedema19 and preserve ATP levels20 in immature rat pups. Allopurinol appears to have an effect as a free radical scavenger as well as inhibiting xanthine oxidase.
Conclusions
Neuronal protection following birth asphyxia by means of pharmacological agents remains a controversial area. There is now little doubt that excessive Ca2+ accumulation within the neuron is an important factor in contributing to irreversible neuronal injury but it is by no means certain that blockade of voltage or agonist calcium channels will improve outcome in the newborn following birth asphyxia. In perinatal animal models, the most promising interventions in brain protection are the NMDA receptor antagonists and a clinical intervention study with magnesium sulphate will soon commence. The CCBs, although associated with potentially severe cardiovascular complications, may also be important drugs particularly when highly specific cerebral T-type blockers become available for human use. In view of early calcium entry being associated with depolarization before AOCCs open, a rational strategy may be to use a combination of both CCBs and NMDA receptor antagonists.
Vascular injury causing impaired reperfusion after asphyxia may also be an important factor in neuronal death and this is most likely to be mediated through generation of free radicals with damage to the cerebrovascular endothelium. Allopurinol appears to be the most effective agent in reducing free radical generation in the brain. A major disadvantage of allopurinol in clinical use is that the intravenous preparation has a pH of 10 which makes it a potentially dangerous solution to give into the umbilical artery or vein and tissue injury will occur if the infusion extravasates into subcutaneous tissues.
References
- McDonald JW, Roeser NF, Silverstein FS, Johnston MV. Quantitative assessment of neuroprotection against NMDA-induced brain injury. Exp Neurol 106:289-296, 1989.
- McDonald JW, Silverstein FS, Cardona D et al. Systemic administration of MK-801 protects against N-methyl-D-aspartate and quisqualate- mediated neurotoxicity in perinatal rats. Neuroscience 36:589-599, 1990.
- McDonald JW, Silverstein FS, Johnston MV. Neuroprotective effects of MK-801, TCP, PCP and CPP against N-methyl-D-aspartate induced neurotoxicity in an in vivo perinatal rat model. Brain Res 490:33-40, 1989.
- Hattori H, Morin AM, Schwartz PH et al. Posthypoxic treatment with MK-801 reduces hypoxic-ischemic damage in the neonatal rat. Neurol 39:713-718, 1989.
- Ford LM, Sanberg PR, Norman AB, Fogelson MH. MK-801 prevents hippocampal neurodegeneration in neonatal hypoxic-ischaemic rats. Arch Neurol 46:1090-1096, 1989.
- Wolf G, Keilhoff G, Fischer S, Hass P. Subcutaneously applied magnesium protects reliably against quinolinate-induced N-methyl-D-aspartate (NMDA) mediated neurodegeneration and convulsions in rats: Are there therapeutical implications? Neurosci Let 117:207-211, 1990.
- Akaike N, Kostyuk PG, Osipchuk YV. Dihydropyridine-sensitive low threshold calcium channels in isolated rat hypothalamic neurones. J Physiol 412:181-91, 1989.
- Takahashi K, Akaike N. Calcium antagonist effects on low-threshold (T-type) calcium current in rat isolated hippocampal CAl pyramdial neurons. J Pharmacol Exp Ther 256:169-75, 1991.
- Alps BJ. Drugs acting on calcium channels: Potential treatment for ischaemic stroke. Br J Clin Pharmac 34:199-206, 1992.
- Noronha-Dutra AA, Steen-Dutra EM, Woolf N. An antioxidant role for calcium antagonists in the prevention of adrenaline mediated myocardial and endothelial damage. Br Heart J 65:322-325, 1991.
- Thiringer K, Hrbek A, Karlsson K et al. Postasphyxial cerebral survival in newborn sheep after treatment with oxygen free radical scavengers and a calcium antagonist. Pediatr Res 22:62-66, 1987.
- Gunn AJ, Mydlar T, Bennet L et al. The neuroprotective actions of a calcium channel antagonist, flunarizine, in the infant rat. Pediatr Res 25:573-576, 1989.
- Silverstein FS, Buchanan K, Hudson C, Johnston MV. Flunarizine limits hypoxia-ischemia induced morphologic injury in immature rat brain. Stroke 17:477-82, 1986.
- Ment LR, Stewart WB, Duncan CC, Pitt BR. Beagle pup model of perinatal asphyxia: Nimodipine studies. Stroke 18:599-605, 1987.
- Levene MI, Gibson NA, Fenton AC et al. The use of a calcium-channel blocker, nicardipine, for severely asphyxiated newborn infants. Develop Med Child Neurol 32:567-574, 1990.
- Zweir JL, Kuppusamy P, Lutty GA. Measurement of endothelial cell free radical generation: Evidence for a central mechanism of free radical injury in postischemic tissues. Proc Nat Acad Sci USA 4046-4050, 1988.
- Siesjo BK, Agardh CD, Bengtsson F. Free radicals and brain damage. Cereb Brain Metab Rev 1:165-211, 1989.
- Goplerud JM, Mishra OP, Delivoria-Papadopoulos M. Brain cell membrane dysfunction following acute asphyxia in newborn piglets. Biol Neonate 61:33-41, 1992.
- Palmer C, Vannucci RC, Towfighi J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res 27:332-336, 1990.
Discussion
William W. Hay, Jr., M.D.
University of Colorado Health Sciences
Center, Denver
Conventional Treatment Interventions
Dr. Lemons focused on the impact of neonatal resuscitation and early medical management procedures. Over the past 20 years, these have become reasonably common and standardized through established programs such as NALS (American Heart Association/American Academy of Pediatrics). While there is general consensus that these procedures have improved the care of the newborn infant, particularly by improving the technical resuscitation knowledge and skills of medical personnel not accustomed to frequent resuscitation opportunities, there is little data to measure their impact. Thus, while few would argue with the importance of intubation to secure the airway, clearing the airway of debris such as meconium, ventilating the lungs, perhaps even moderately hyperventilating to ensure lung expansion and to relieve respiratory acidemia, administering oxygen, performing chest massage and infusing blood volume expanders when circulation definitely is inadequate, etc., no controlled clinical trials, random or otherwise, have proven their efficacy with respect to preventing or reducing the severity of HIE (asphyxial brain injury). Some old standbys could be questioned in relation to the potential benefit of newer treatments such as drying and warming versus cerebral hypothermia (particularly via the circulation). Some previously-established therapies such as the administration of sodium bicarbonate should be discontinued or at least reserved for carefully controlled attempts to relieve persistent, severe metabolic acidosis—although there is reasonable and serious concern that even this aspect of bicarbonate treatment is more deleterious than beneficial.1 Furthermore, while there has accumulated some limited evidence that lactic acidosis may be harmful, it is best treated by restoring normal oxygenation and perfusion; pure metabolic acidosis (e.g., as produced by infusion of hydrochloric acid) has not proven damaging in animals, even to very low pH levels (6.8-7.0). And there still is variety in treatment, such as the use of different concentrations of oxygen, how early in the course and how quickly intervention is provided, who are involved as the care providers, the degree of salt and water restriction, the levels of blood pressure, P02, PCO2, and pH to maintain, the administration of anticonvulsants (phenobarbital is most commonly used, but there is considerable debate about its effects and there is little consensus on others), steroids, hyperosmotic agents, the influence of and need to treat hyperbilirubinemia, and the initiation, type, route of administration, and amount of nutrition. Of particular note is the unresolved debate about the potential benefit vs. harm produced by infusion of glucose and mild to moderate hyperglycemia. Clearly, adult studies have produced contradictory results compared with neonatal studies. At least normoglycemia is considered appropriate. Whether such variation in one or a combination of these interventions will affect outcome is not known.
Conventional interventions during labor have focused on administration of oxygen, maternal positioning, and emergency delivery, usually by cesarean section. These interventions are usually in response to abnormal fetal heart rate tracings and/or scalp blood gas analysis. A major difficulty with such clinical guidelines is that they provide limited accuracy in predicting the actual impairment of the fetus at the time or what will happen if one form or another of intervention is applied.
In both intrapartum and postpartum situations, when fetal or neonatal asphyxia is presumed to have occurred or to be occurring, conventional interventions over the past 25 years have improved general supportive care of infants but their measurable impact on improving the outcome of perinatal asphyxia is limited. On the other hand, there seems little that will or can be given up of these conventional interventions. Whether further testing is needed remains debatable but this is unlikely to produce major new knowledge or advances in clinical management and outcome.
Neuronal Rescue and Prophylaxis: Studies in Animals
Dr. Gluckman focused on animal studies. Data collected from a variety of animal models shows that their comparability to each other is variable and uncertain, and their representation of human asphyxia is similarly not proven. This is in large part because there is insufficient data in humans, especially the fetus, to provide direct comparison of physiological and biochemical parameters during experimental and clinical asphyxial episodes. Much greater capacity to accurately measure on line physiological and biochemical values in fetuses and newborn infants is required and is essential if animal studies are to be successfully extrapolated to human interventions, and especially to understand what the results are in human infants. Furthermore, some models are well studied and are even reproducible, but their relation to human clinical conditions uncertain; e.g., the carotid artery ligation model is a commonly used experimental animal model but no human infant (except perhaps for those infants placed on ECMO) suffers this pathological process. Furthermore, effects of hypoxia, ischemia, and acidosis, the stage of neuronal development, the presence and effects of confounding variables (drugs, prior medical problems such as undernutrition, etc.) the nature, duration, severity etc. of the pathological processes, etc., are not uniform among cases of asphyxia or among models and this may reflect variability in responses to certain interventions as well as neonatal outcome.
Regarding prophylactic experimental drugs (gangliosides such as GM 1, lazeroids or 21-amino steroids) which appear to function by stabilizing neuronal membranes, their administration to the fetus will require knowledge that asphyxia is going to occur. This is easy to predict (by control) in an experimental model but not at all easy in a human infant. Again, access to the fetus to measure responses is needed, not just more fetal heart rate tracings. If administration to the mother is the route of getting the drug to the fetus, then placental transport function ought to be known or at least normal—currently there are no good measures clinically of placental transport of these substances. These same issues and concerns apply to the other interventions discussed, including glutamate antagonists (NMDA receptor and kainic acid receptor antagonists), free radical scavengers, and free radical synthesis inhibitors, as well as cerebral hypothermia. In all cases, the developmental state of the brain and the presence and effects of modifiers must be understood and measurable. Thus, although the results of some early trials in animal models are encouraging, there is considerable basic and applied research yet to do before clinical trials can begin.
Regarding rescue treatments, immune modulation with TGFb, anticonvulsants (MK801), neurotrophins (IGF-I), and apoptosis inhibition (IGF-I, IGF binding proteins, other growth factors, protein synthesis inhibitors) were discussed. Use of these substances requires knowledge of when the asphyxia occurred and experimental evidence of their effects at different times following the onset or end of an asphyxial episode. This is information more easily obtained from experimental than clinical trials.
If nothing else, this review pointed to critical areas for future investigation of potential therapies, including timing of administration relative to the insult, relative to the stage of maturation of the infant, and relative to the metabolic, thermal, and cardiovascular status of the infant. In all cases, side effects of the drugs and interventions are only minimally understood, in animal models or in human infants.
Experimental Treatments in Humans
Dr. Levene focused on experimental treatments in humans using glutamate antagonists, calcium channel blockers and allopurinal. NMDA receptor antagonists function as glutamate antagonists and prevent the excessive calcium influx into affected neurons. The gestational age of infants under study or treatment is important as the immature brain appears to have proportionately more NMDA receptors than the mature brain.2 Timing of administration is important as is the hierarchy of drug effects, as the effects of competitive inhibitory and enhancing substances are not well known.3 Magnesium seems one of the more promising agents in this area of inhibiting calcium uptake by affected neurons as it has a large margin of safety and there is considerable clinical experience with its administration to fetuses and newborn infants via the mother.4 In contrast, most calcium channel blockers have marked and potentially deleterious side effects, particularly systemic hypotension.5 Allopurinol blocks xanthine oxidase formation and production of free radicals.6 Its pharmaceutical preparation has a pH of 10-11. making its clinical application potentially dangerous.
Obviously, clinical trials have been quite limited as investigators have been trying to understand the role of various substances and interventions in more controlled circumstances. As pointed out above, it remains essential to develop much better capacity to measure fetal and neonatal physiology and biochemistry accurately and on line so that the timing of events and the responses to pathology and interventions can be observed and measured. These problems remain the major stumbling blocks to further clinical work. It also is unfortunate that clinical trials in the look see stage get more notoriety than they deserve, often well before development of hard and reproducible data from randomized, controlled clinical trials.
Conclusions
- More basic knowledge is needed of the pathophysiolgical and biochemical processes producing and occurring with fetal and neonatal asphyxia, and the status (developmental stage, physiological condition, etc.) of the infant when the insult occurs.
- The means to measure the affected and critical physiological and biochemical disturbances are critically needed.
- There must be more access to the fetus, especially for on line data.
- This is an exciting time because there is much more data about how asphyxial injury occurs, and there are many new pharmacological and physiological interventions available, so that we can, with better rationale, treat more babies, more often. We need ways to know what to treat, when to treat, and how to observe and measure the effects of treatment on the pathophysiology that we see and know causes injury.
- Proof of comparability of animal models and human fetal and neonatal asphyxial conditions.
- Knowledge of side effects of interventions.
- Knowledge of interactions of interventions and potentially confounding variables (e.g., gestational age, prior insults, other drugs, nutritional state, etc.)
- More research on dose-response relationships, side effects, interactions.
- For prophylaxis, how does one know when an insult is going to occur?
- For rescue, when did the insult occur, how long did it last, what was it, what did it do, how bad was it?
- We need more information about placental function and the role of the placenta in the asphyxial process, as well as the functional capacity to administer treatments to the fetus via the placenta from the mother. We also need more ways to treat placental dysfunction.
- We need to know how fetuses and babies with pre-existing CNS pathology act during normal as well as pathological labor, and in the presence of hypoxia and ischemia.
Final Comment
It is unlikely that we will have much impact on the outcome of infants suffering from hypoxia- ischemia (or asphyxia) if we wait until we see it, in spite of strides in the development of rescue interventions.
We certainly do not allow babies or mothers to get so sick before we prevent further abnormal pathophysiological development. Certainly we owe the fetus the same respect and treatment.
References
- Tyson JE. Immediate care of the newborn infant. In: Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: Oxford University Press, pp. 21-39, 1992.
- McDonald JW, Roeser NF, Silverstein FS, Johnston MV. Quantitative assessment of neuroprotection against NMDA-induced brain injury. Exp Neurol 106:289-296, 1989.
- McDonald JW, Silverstein FS, Cardona D et al. Systemic administration of MK-801 protects against N-methyl-D-aspartate and quisqualate- mediated neurotoxicity in perinatal rats. Neuroscience 36:589-599, 1990.
- Wolf G, Keilhoff G, Fischer S, Hass P. Subcutaneously applied magnesium protects reliably against quinolinate-induced N-methyl-D-aspartate (NMDA) mediated neurodegeneration and convulsions in rats: Are there therapeutical implications? Neurosci Let 117:207-211, 1990.
- Akaike N, Kostyuk PG, Osipchuk YV. Dihydropyridine-sensitive low threshold calcium channels in isolated rat hypothalamic neurones. J Physiol 412:181-91, 1989.
- Takahashi K, Akaike N. Calcium antagonist effects on low-threshold (T-type) calcium current in rat isolated hippocampal CAl pyramidal neurons. J Pharmacol Exp Ther 256:169-75, 1991.
Discussion
Michael V. Johnston, M.D.
Department of Neurology and Pediatrics, Johns
Hopkins University School of Medicine and
Kennedy Krieger Institute, Baltimore, Maryland
Interventions Aimed at Preserving Brain Tissue Injured by Hypoxia-ischemia
In a little more than a decade, it has become plausible to discuss interventions aimed at preserving brain tissue injured by hypoxia-ischemia. Conceptually, we have moved from a theory of injury based primarily on a mismatch between energetic demands and energy delivery to a model of a cascade of self-destructive calcium-mediated events triggered by hypoxia-ischemia. Previous pharmacologic therapies based on reducing the brain's energy demands, such as barbiturates are largely ineffective. It has been demonstrated in animals that the interruption of several key steps in this auto-destructive cascade can protect and salvage brain tissue. These excellent papers reflect the important shifts in our thinking about the problem of perinatal asphyxia. Demonstration that drugs can protect the brain from hypoxic-ischemic injury has given new energy to this field because the hope that a useful intervention for humans can be identified.
The most attractive therapies are both robustly active and based on an understandable mechanism. If we evaluate each of the therapies that have been shown to have some effect in animal models the glutamate antagonist drugs emerge as the most promising. Investigators who had experience with emerging therapies such as voltage sensitive calcium channel antagonists recognized the effects of NMDA glutamate antagonists such as MK-801 as a quantum jump in neuroprotective activity. Hundreds of publications have described the neuroprotective effects of MK-801 and its cousins in a variety of models of hypoxia-ischemia. This receptor directed strategy is all the more attractive because it is linked to an understanding of the special importance of glutamate receptors in the developing brain.1 Glutamate receptors, especially NMDA receptor channel complexes, appear to have important roles to shape developing neuronal networks. For example, in the developing visual cortex of kittens NMDA mediated visual evoked responses are enhanced compared with the response in adults. Blockade of these receptors diminishes the ability of electrical activity to shape cortical ocular dominance columns. The immature NMDA receptor channel complexes have pharmacologic, molecular and electrophysiologic characteristics that allow them to play an enhanced role in the immature brain. In addition, the opening of the channels is cleverly enhanced by energy deficiency through the receptor's unique magnesium blockade characteristics. The emerging evidence indicates that the NMDA receptor channel complexes in neonates compared with the adult are activated in order to serve a temporary but vital developmental role.
Hypoxia-ischemia is especially likely to disrupt neurons that bear these channels by delivering a triple whammy—enhanced glutamate released from nerve terminals, paralyzed re-uptake of glutamate into nerve terminals and opening channels when magnesium is removed during energy deficiency. According to the information available on the development of these systems, parts of the brain that are making maximal developmental use of these receptors at the time of hypoxia-ischemia are most vulnerable to death. The theory supports the contention that patterns of selective vulnerability in the developing brain can be understood according to the distribution of glutamate receptors as well as to changes in regional cerebral blood flow.
The glutamate hypothesis of injury is also appealing because it is compatible with well-known but sometime puzzling clinical features of hypoxic-ischemic encephalopathy. One of these features is that severe hypoxia-ischemic encephalopathy is often associated with hyperexcitability or seizures. Prior to the glutamate hypothesis, it was difficult to understand how a disorder produced primarily by energy deprivation led to excitatory signs. What we have learned about excitatory neuron transmitter systems indicates that the neonatal brain is probably skewed towards excitability compared with the adult and that the delicate mechanisms that control this level of excitability are energy sensitive. The theory is also consistent with the threshold phenomenon seen in hypoxic-ischemic disorders. Hypoxia, often of fairly severe degree, can be tolerated well by the infant nervous system while cerebral blood flow increases to compensate. We can envision that the energy dependent pumps that control glutamate uptake and calcium homeostasis within the neuron can be maintained even under severe stress but when a threshold of ischemia is reached the synapse becomes flooded with glutamate and the flood gates of the NMDA channels become stuck open.
Another attractive, though unproven, hypothesis linking basic science to clinical observation, is that the special sensitivity of the motor system to injury which produces the cerebral palsy syndrome is related to activated excitatory mechanisms used by these systems in the perinatal period. It can be speculated that neuronal systems undergoing a peak of plasticity might have greater activation of NMDA receptors and therefore be more vulnerable to injury. During the period from full term birth throughout the first year the motor systems in the brain might be considered as among the most rapidly changing. An analogy can be made with the sensitivity of computers to power surges produced by a strike of lightning. Like electrons which burn out delicate electronic chips, the charge-carrying calcium ions trigger autolysis for the delicate intracellular machinery of developing neurons.
Although glutamate blockers are the most promising agents, other compounds already mentioned may emerge as promising therapies that could surpass the glutamate blockers. However some, such as voltage sensitive calcium antagonists, are far less potent than the glutamate blockers and produce troublesome systemic effects. Others such as the gangliosides appear to rival them in some models but their mechanisms is poorly understood. Similarly, protein growth factors may have an important effect to rescue injured tissue. However, the complexity of their actions, their expense and the barrier to answering questions about the nervous system make their use more speculative.
If glutamate antagonists are the drugs of the moment and are the best current candidates for clinical trials, how should we proceed? The most daunting impediment to clinical usage is the primary topic of this conference, the definition of perinatal asphyxia and the definition of a clinical population worthy of intervention. We need to define the markers of tissue hypoxia-ischemia that are highly associated with tissue loss. Defining a high-risk group with a predictable outcome without therapy would greatly aid these efforts. This is especially important so as not to expose too many normal infants to drugs with potential adverse effects. Even with a careful definition, however, variability in outcome would remain a problem for a study design.
I would like to suggest two kinds of clinical studies that might be used to put a candidate glutamate antagonist to the test. One study I would propose would aim to determine if a drug stopped seizures and helped normalize the EEG in patients with hypoxic-ischemic encephalopathy This concept which was suggested for a trial of phenobarbital, is based on the hypothesis that a glutamate blocking drug that blocks seizures from hypoxia-ischemia might use similar mechanisms to preserve tissue. For this study, a fairly homogenous group could be defined using video/EEG monitoring in the nursery.
Another study group might involve babies undergoing cardiac surgery, especially the group subjected to hypothermic/circulatory arrest. This would be a somewhat older group than the perinatal asphyxia group but the advantage would be that prospective assessment of each infant prior to surgery as well as prospective drug treatment would be possible. In studies of adult animals subjected to hypothermic circulatory arrest and conducted with collegeaus in cardiac surgery both MK-801 and GM 1 gangliosides block selective neuronal necrosis and neurologic dysfunction.2 Use of drugs in this rather controlled setting might provide information about effects and toxicity prior to trials in asphyxiated infants especially those who are still in utero.
Choice of glutamate antagonists for these trials needs to be made carefully and collaboratively Some of the NMDA channel antagonists such as ketamine and PCP are potent anesthetics while MK-801 appears to have less effect on respiration. Interestingly the non-NMDA antagonists including CNQX and DNQX appear to be potent respiratory toxins probably because glutamate is used in respiratory control centers in the brainstem. MK-801 has been used safely in patients with epilepsy and status epilepticus. Based on behavioral studies, drugs that antagonize the competitive NMDA site such as CGS-19755 may have fewer cognitive side effects. This drugs is now undergoing early human trials for head injury in Glasgow. The cough suppressant dextromethorphan is a moderately potent NMDA channel blocker and has shown activity to protect against hypoxia-ischemia in animals. Several infants with the metabolic disorder, hyperglycinemia have been treated with dextromethorphan to successfully block the seizures and EEG abnormalities present in this disorder. A case that we have treated is now several years old.3 Another promising approach may be to block the glycine site on the NMDA receptor channel complex. Several glycine antagonists (not antagonists of the inhibitory strychnine site) are potent nerve protective drugs and the recently approved anticonvulsant felbamate has been shown to have potent neuroprotective effects.4 This drug might be a potential choice for a trial against hypoxia-ischemic seizures in newborns.
In summary, these excellent papers illustrate how far we have come in thinking about a disorder which has traditionally been untreatable. We now have a variety of approaches and protocols to choose from.
References
- McDonald JW, Johnston MV. Physiological and pathophysiological roles of excitatory amino acids during central nervous system development. Brain Res Rev 15:41-70, 1990.
- Blue ME, Redmond JM, Zehr K et al. Altered excitatory amino acid receptor expression after hypothermic circulatory arrest in the dog. Society for Neuroscience Abstracts, 1993.
- Hamosh A, McDonald JW, Valle D et al. Dextromethorphan and high dose benzoate therapy for nonketotic hyperglycinemia in an infant. J Pediatrics 121:131-135, 1992.
- Wasterlain CG, Wallis RA, Adams LM, Panizzon K. Felbamate is a potent neuroprotective agent. Epilepsia 34, Suppl 2, p 92, 1993.
Session V: Clinical Studies of Long-Term Outcome
Moderator: John Freeman
Long-Term Outcome in Birth Asphyxia—The Role of Neonatal Encephalopathy in Prediction
Karin Nelson, M.D.
Neuroepidemiology Branch (NEB),
National Institute of Neurological
Disorders and Stroke (NINDS),
National Institutes of Health,
Bethesda, Maryland
Introduction
The long-term outcome of birth asphyxia that is of central interest to a neurologist, and probably to most others including parents, is neurologic status. In asking the long-term neurologic outcome of birth asphyxia, we encounter a number of problems. First, there is no generally available means for the objective measurement of birth asphyxia, allowing surety as to whether nonspecific clinical signs in the neonate are appropriately attributable to asphyxia. Second, we have far too few good studies of the natural history of birth asphyxia, studies in defined populations that were not selected for factors that might have an impact on outcome, that included clear and reasonable criteria for asphyxia, that followed the studied children to a time when some reasonably homogeneous outcome condition could be reasonably reliably ascertained, and that were large enough to permit conclusions about relationships between uncommon events and uncommon outcomes.
In addition to these difficulties and a number of others, we have probably been compounding our problems by using the same words, birth asphyxia, to mean a number of different things. Birth asphyxia occurs in a wide range of degrees, from the normal low oxygen tension of the intrauterine environment and of birth, to acute total and prolonged absence of gas exchange. The same words have in general been employed for all of these. Crudely diagrammed (Figure 1), the outer boundary of birth asphyxia, BA-I, represents some degree and duration of asphyxia differing from whatever norms are employed.
FIGURE 1: Degrees of Birth Asphyxia and Their Relationship to Outcome
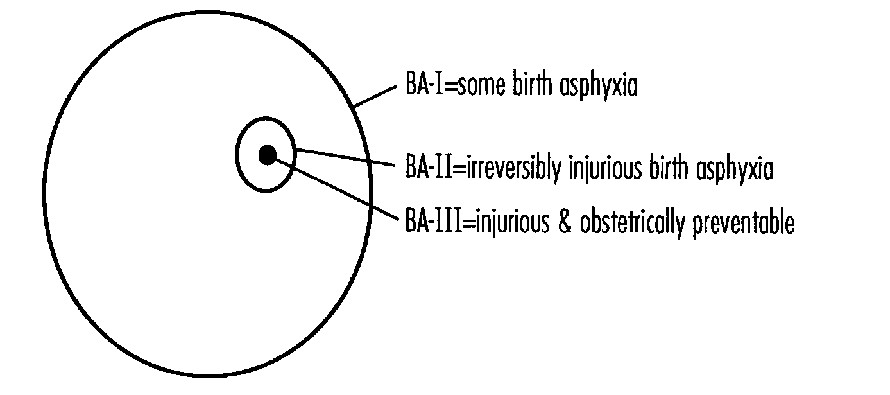
The outer circle, BA-I, represents the presence of some degree of birth asphyxia. BA-II, a subset, represents the degree and duration of birth asphyxia required to produce irreverisble brain injury in a term fetus/neonate. BA-III, the size of which relative to the others is not known, represents the degree and duration of brith asphyxia required for irreversible brain injury, and obstetrically preventable.
BA-I may be useful to obstetricians and to neonatologists as a signal of need for increased vigilance lest problems progress. The second circle, BA-II, represents the degree and duration of asphyxia that is associated with a substantial risk of irreversible brain injury in the term fetus who survives it. BA-II is a subset, and a fairly small subset, of BA-I, as indicated by repeated observations that in term infants, whatever definitions the various studies have employed, the majority of survivors of even severe and lengthy asphyxia are later clinically normal.1-3 One reason interventions designed to prevent damage from birth asphyxia have apparently not been associated with lower rates of perinatal mortality or of later neurologic morbidity may be that the monitoring methods have identified BA-I and not BA-II, resulting in interventions directed largely at mother-baby pairs in whom there would not have been an excess of long-term morbidity even without the intervention. (Other possibilities have been discussed by Grant.)4 A third circle, BA-III, its relative size entirely unknown, is a subset of BA-II, and represents the degree and duration of birth asphyxia that is injurious to brain and is obstetrically preventable.
FIGURE 2: Asphyxia Associated with Long-term Adverse Outcome Only When Neonatal Encephalopathy is Present
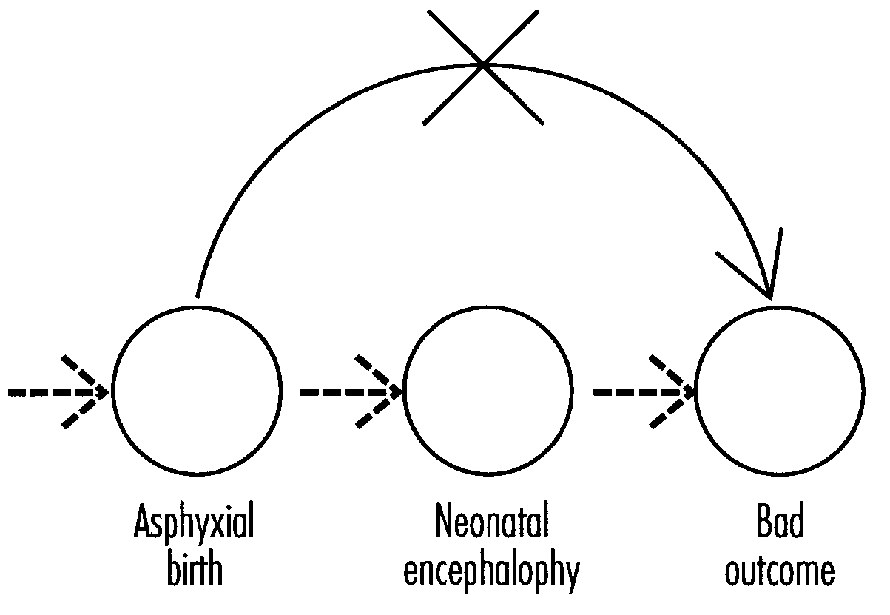
Another range of differences encompassed by our use of the words birth asphyxia has to do with when in the course of illness a defect of gas exchange occurs. Some infants thought to be asphyxiated, perhaps with demonstrated poor cerebral perfusion, are born following such obstetric catastrophes as placental abruption, cord prolapse with lengthy occlusion, or maternal shock. More often no such clearcut obstetrical antecedent is identified. We have no convincing answers as to why these infants are affected. It is probably not reasonable to assume without investigation of other possible risk factors that the initiating pathology must have been acutely asphyxial in all or most such cases, since many pathogenetic mechanisms trigger changes in the biology of cells that, once they are underway, can result in ischemia as a late result. For example, Leviton5 has recently noted that tumor necrosis factor, associated with infection and inflammation, can produce hypotension which can in turn lead to ischemia; he has raised the possibility that agents such as tumor necrosis factor and other cytokines, known to play a role in triggering preterm birth, might themselves contribute to brain pathology in prematurely born infants, or in those born at term. Leviton's focus was on factors that might contribute to injury to white matter, but experimental evidence also suggests that certain cytokines can affect neurons in developing brain in exceedingly low concentrations, the effects differing by developmental stage.6 Whether or not these interesting hypotheses prove correct, the point for this discussion is that ischemia is a late common path for many kinds of illnesses. Ultimately most of us die of asphyxia, in a sense; is it a medically interesting sense?
Ignoring the chain of events that precedes hypoxia or ischemia may prevent focus on real and important differences, and may discourage needed clinical research such as, for example, investigating whether certain maternal or pregnancy conditions, including infectious or inflammatory illnesses, predispose to hypotension or cerebral hypoperfusion in the term or preterm neonate.
Once hypoxemia or poor cerebral blood flow is identified in the infant, consideration of trials of therapy may be warranted in an attempt to interrupt the chain of damage whatever the cause. However, therapy at that point is damage control; if we are to develop better strategies for primary prevention, it is necessary that we learn to identify the instigating pathology. The implications are quite different if ischemia is the end stage of a process that began with chorionitis or immune disturbance, for example, than if the defect in gas exchange arose by way of placental abruption or maternal shock. Using the words birth asphyxia and ischemia for these quite different situations may also have unwarranted implications concerning responsibility and blame.
The difficulties we face in approaching questions of the relationship of birth asphyxia to outcome are real and formidable. Using the same words to mean a range of different things is one part of the problem that we can and should avoid.
What Prenatal or Neonatal Factors that Might Be Related to Birth Asphyxia Predict Long-Term Neurologic Outcome?
Consideration is limited to term singleton infants (free of major malformations), because for low birthweight infants or those multiply born other issues may arise, and also because evidence is even more sparse, selected, and heterogeneous than for term singletons.
What long-term neurologic outcomes have we have reason to think might be related to birth asphyxia? Sameroff wrote, "The intellectual outcomes for children (mental retardation and learning disorders) were far better explained by the small set of family factors than on any combination of the multitude of biomedical variables."7 Paneth recently noted,8 "A very consistent finding in the literature is that when adverse perinatal events are implicated in the causation of a neurodevelopmental disorder, cerebral palsy is invariably present." Current evidence indicates that the outcome to track in studying the long-term outcome of birth asphyxia is cerebral palsy (CP), which may be accompanied by mental retardation, seizure disorder, or other associated neurologic or sensory disability.
Obstetric Factors
Three obstetric conditions especially linked with asphyxial birth are placenta previa, abruptio placentae, and prolapsed umbilical cord. All these conditions are uncommon, the most common of them, abruption, occurring in 1.7 percent of births.9 All these conditions are associated with increased risk of preterm delivery, so they are even less common in term births. In a cohort of more than 50,000 live births, only one of these conditions—placenta previa—was associated in infants over 2,500 g with a risk of CP that significantly exceeded the risk in children whose births were uncomplicated, and the absolute magnitude of risk in term survivors was less than 2 percent.9 These asphyxial obstetric conditions were not strikingly powerful as predictors of long-term neurologic deficit.
Furthermore, even these seemingly acute obstetric catastrophes have histories that may go far back into pregnancy. Placenta previa is obviously determined at the time of implantation. If there are disadvantages to the fetus who has his or her placenta implanted in the isthmic area of the uterus rather than in the fundus, then those disadvantages have been present in pregnancies with placenta previa for a long time before labor begins. Placenta previa is associated with higher rates of spontaneous abortion, with male fetuses, advanced maternal age, parity, impaired fertility, smoking, and developmental defects of the fetus.10,11 Placenta previa may be recurrent in the same gravidae.10 Whether the increase in risk of cerebral palsy associated with placenta previa is fully preventable by obstetric interventions at the time of birth is not known.
Abruption too may have early precursors including maternal age, smoking history, cocaine exposure or frequent use of marijuana, preeclampsia, chorionitis, and fetal malformation.12, 13 Cord prolapse is often associated with breech birth, which is disproportionately often associated with conditions characterized by weakness or hypotonia in the fetus.
So even previa, abruption and cord prolapse were not strikingly good as predictors of CP, and these conditions are not necessarily bolt-from- the-blue acute hypoxic or ischemic events that begin during labor or delivery. These factors have their own histories, some of which have implications for development. Although all of these can be associated with fetal hypoxemia, the relationship of each with fetal neurologic condition may be considerably more complicated than the words birth asphyxia imply.
Neurologic Signs
Among signs apparent in the newborn immediately after birth that might be evaluated as predictive of long-term outcome are those recorded in the Apgar score. Low Apgar scores are indeed predictors of marked increase in risk of CP in term survivors, but only at the rare extreme: it is very low scores (3 or less) very late (after 10 minutes) that are strongly predictive.2 These very low late scores are very uncommon and when they do occur are often not survived.2 Nevertheless, extremely deviant Apgar scores have high positive predictive value for later CP in survivors.
Do low Apgar scores identify asphyxial birth? Very low late scores, those that are associated with adverse outcome, might have antecedents different from factors that predispose to transient mild depressions of Apgar score; I know of no examination of this possibility.
Hypotonia, respiratory delay, difficulty in maintaining respiration, and poor reflex responsivity all are or can be evidence of neurologic dysfunction. When such signs of neurologic depression are recognized in the delivery room they are called low Apgar scores. The same signs identified in the neonatal nursery (Table 1), with or without the addition of seizures and with some variously defined exclusions, are often referred to as hypoxic-ischemic encephalopathy. Low Apgar scores are not specific to any etiology, and neither is a constellation of similar signs observed over the next days of life. Defining birth asphyxia by Apgar scores alone is no longer acceptable.
Neurologic signs that appear or persist after the first minutes or hours of life are also predictors of neurologic outcome. Indeed, obstetrical complications were predictive only if other neurologic signs were present in the neonatal period (Table 2).14 Subdivided to give special attention to neonatal seizures, these factors show a striking relationship with rate of later CP (Table 3).15 Obstetrical complications or low Apgar scores were related to CP risk only if followed by neonatal encephalopathy, and then there was dramatic increase in risk. Children with that combination of ominous predictive factors were 0.06 percent of term infants.15 They contributed 16 percent of the CP in that population.
TABLE 1: Similarities: Low Apgar Score and Hypoxic-Ishemic Encephalopathy
| DELIVERY ROOM | NURSERY | |
|
Hyptonia |
||
| LOW APGAR SCORE | HYPOXIC-ISCHEMIC ENCEPHALOPATHY |
Levene et al.16 found low Apgar scores to predict CP, but reported that "No handicapped children were found who had had low Apgar scores alone without encephalopathy." Hadders-Algra et al.17 made a similar observation: "The Apgar score was the sole obstetrical variable which contributed significantly to an increased risk of neurological handicap, but only in the presence of neonatal (neurologic) deviancy."
Klipstein and McBride18 noted that number of days on respirator, days to full oral feeds, and days to normal level of activity were predictive of CP in term newborns they considered asphyxiated at birth. Clearly, all these are measures of presence and severity of neurologic disturbance.
A substantial body of evidence indicates that in the term baby, if a complicated birth is not followed by neurologic symptomatology, and usually by symptomatology also in other organ systems, then there is no increase in risk of CP. Asphyxial birth is not associated with heightened risk of unfavorable neurologic outcome unless there is neonatal encephalopathy (Figure 2). The pathway from asphyxial birth to long-term bad outcome is via neonatal encephalopathy.
A major predictor of long-term neurologic disability, then, is short-term neurologic abnormality, neonatal encephalopathy. Evidence from the National Collaborative Perinatal Project suggests that neonatal depression present from birth and with continuing manifestations that culminate in neonatal seizures is a particularly dire predictor.15 This cluster of factors needs evaluation in a current dataset.
Low Apgar scores and low pH account for only a small minority of encephalopathy in term babies.19 There are obviously causes of neonatal encephalopathy other than asphyxial birth and some or many of these may evade our short list of exclusions. For CP we have some ideas about other possible causes,20,21 but we have only scattered and nonconvergent ideas of what these factors are. Can infection and the associated lactic acidosis predispose to encephalopathy in the neonate? If the mother or placenta are infected, can factors associated with infection such as cytokines reach the fetus and affect it even if the fetus is not itself infected? There is great need for clinical examination of hypotheses derived from the experimental neurosciences.
The only study I know of to examine a whole range of clinical factors as possible antecedents of neonatal encephalopathy in term infants is an investigation now underway in the Western Australia Research Institute for Child Health. Evidence from the pilot phase of that study suggests some surprises: births in neonates with encephalopathy were not strikingly different in most regards from births of controls, but administration of thyroid hormone to mothers and maternal fevers in labor were observed in significant excess in infants who manifested encephalopathy as neonates as compared with controls.
Most CP, and virtually all of mental retardation, learning disorders, and so on, in children who do not also have CP, has causes other than birth asphyxia. So, it appears likely, does neonatal encephalopathy. The contribution of these other predictors of neonatal encephalopathy to later neurologic disability remains to be determined in studies that investigate rather than assume etiology.
TABLE 2: Obstetric Complications and Cerebral Palsy Rate Related to Signs of Neonatal Encephalopathy in Infants over 2500 g.14
| CEREBRAL PALSY RATE PER 1000 | ||
| Obstetric complications | No encephalopathy | 3 + signs of encephalopathy |
|---|---|---|
| no | 2.4 | 70 |
| yes | 2.3 | 122 |
| yes + 5' Apgar <5 | 3.5 | 269 |
From: Nelson and Ellenberg14
TABLE 3: Neonatal Neurologic Signs as Predictors of Cerebral Palsy in Infants Over 2500 g.15
| 5' Apgar score <5 | Neonatal signs | Seizures | CP/1000 |
|---|---|---|---|
| 0 | 0 | 0 | 13 |
| 0 | 0 | + | 13 |
| + | + | + | 550 |
From: Ellenberg and Nelson15
Acid-Base Measures
Low pH at or soon after birth, especially with a metabolic component, is a factor associated with multiorgan problems including signs of neonatal encephalopathy.23 Selection factors in most existing studies make judging frequency difficult, but this is an uncommon combination: the 98 term children Portman et al.23 considered to be asphyxiated came from 2 years of births in a population base of 60,000 to 75,000 births per year which, assuming a middle-range of 135,000 births, suggests a rate of moderate or severe asphyxia by their criteria of about 0.07 percent in term infants.
Studies starting with a defined denominator have found that pH, with or without Apgar scores, accounts for very little of neonatal encephalopathy: most infants with encephalopathy are not acidotic, and most acidotic—even severely acidotic—infants are not encephalopathic.19 Only weakly predictive of encephalopathy in the newborn period, measures of acid-base balance including umbilical artery pH have not been shown to be good predictors of long-term outcome.24-26 It would seem reasonable to guess, and recent studies suggest,26, 27 that pH may operate as a predictor in a manner similar to that of Apgar scores, predictive chiefly at the rare extremes. If that is so, then there is likely to be a very high rate of false positive identification, and very large studies will be required to demonstrate relationships. An important additional consideration is that other prenatal factors may influence or determine outcome: in two recent studies of infants with pH below 7.00, a majority of those with adverse outcome had prenatal abnormalities.27, 28
Studies of pH have seldom been performed in large and unselected populations in which outcome is systematically evaluated in a manner permitting estimations of magnitudes of risk. Information on the natural history of children with low pH values with or without base deficit and combining that information with Apgar score would be very desirable if we hope to use pH and base deficit, or combinations of acid-base measures and Apgar scores, as important factors in selecting babies for trials of new therapies for birth asphyxia.
Conclusions
- A major predictor of long-term neurologic outcome is short-term neurologic outcome, neonatal encephalopathy. We need substantially more information on the differential diagnosis of this nonspecific clinical syndrome, and more information also on the natural history of its specific subsyndromes.
- Defining birth asphyxia by Apgar scores alone is no longer acceptable. Many definitions of birth asphyxia now proposed include acidosis. This is probably an advance, but acidosis is also not etiologically specific. We need more information on differential diagnosis and natural history of specific combinations of factors proposed for the definition of birth asphyxia, as well as of factors that modify risk. This is especially critical if we are to use these criteria as a basis for selecting babies to receive experimental treatments soon after birth.
- When defective oxygenation is identified or suspected, it may be important to clarify at what point in the clinical course inadequate oxygenation is thought to have arisen.
- The incorporation into perinatology and neonatology of new findings in cell biology and in particular of developmental neurobiology is likely to contribute greatly to our understanding of developmental neurologic abnormalities. Oxygen is not the only molecule important to the developing nervous system. Developmental neurobiology may also help us to anticipate and avoid some unintended harms in trials of experimental therapies.
- Current evidence is that irreversible brain damage from asphyxia during birth in term infants is very uncommon, and that the signs available at the times when decisions must be made as to whether to intervene are low in specificity: the fetus/infant who is truly in trouble may share similar early signs with a very much larger number of other fetuses/infants, the great majority of whom would fare well if they survive without new interventions. In seeking to prevent irreversible brain injury from birth in a small proportion of term infants, we are faced with the possibility that interventions that may save a small number of babies might increase risk for a much larger number. To establish a basis for comparing risks and benefits, as well as costs, we must make wise use of randomized clinical trials.
References
- Scott H. Outcome of very severe birth asphyxia. Arch Dis Child 51:712-716, 1976.
- Nelson KB, Ellenberg JH. Apgar scores as predictors of chronic neurologic disability. Pediatrics 68:36-44, 1981.
- Seidman D, Paz I, Laor A et al. Apgar scores and cognitive performance at seven years of age. Obstet Gynecol 77:875-878, 1991.
- Grant A. Epidemiological principles for the evaluation of monitoring programs—the Dublin experience. Clin Invest Med 16:149-158, 1993.
- Leviton A. Preterm birth and cerebral palsy: Is tumor necrosis factor the missing link? Develop Med Child Neurol 35:549-558, 1993.
- Brenneman DE, Schultzberg M, Bartfai T, Gozes I. Cytokine regulation of neuronal survival. J Neurochem 58:454-460, 1992.
- Sameroff A. Forward. In: Broman S, Bien E, Shaughnessy. Low Achieving Children: The First Seven Years. Hillsdale, NJ: Erlbaum Associates, pp. vii-xi, 1985.
- Paneth N. The causes of cerebral palsy, recent evidence. Clin Invest Med 16:95-102, 1993.
- Nelson KB, Ellenberg JH. Obstetric complications as risk factors for cerebral palsy or seizure disorders. J Am Medical Assoc 251:1843-1848, 1984.
- KelIy JV, Iffy L. Chapter 63. Placenta previa. In: Iffy L, Kaminetzky HA (eds). Principles and Practice of Obstetrics and Perinatology. New York: Wiley & Sons, Vol 2, pp. 1105-1117, 1981.
- Iyasu S, Saaftlas AK, Rowley DL et al. The epidemiology of placenta previa in the United States, 1979 through 1987. Am J Obstet Gynecol 168:1424-1429, 1993.
- Naeye RL. Disorders of the placenta, fetus, and neonate: Diagnosis and clinical significance. St. Louis: Mosby, 1992.
- Williams MA, Lieberman E, Mittendorf R et al. Risk factors for abruptio placentae. Am J Epidemiol 134:965-972, 1991.
- Nelson KB, Ellenberg JH. The asymptomatic newborn and risk of cerebral palsy. Am J Dis Child 141:1333-1335, 1987.
- Ellenberg JH, Nelson KB. Cluster of perinatal events identifying infants at high risk for death or disability. J Pediatr 113:546-552, 1988.
- Levene MI, Grindulis M, Sands C, Moore JR. Comparison of two methods of predicting outcome in perinatal asphyxia. Lancet 1:67-68, 1986.
- Hadders-Algra M, Huisjes HJ, Touwen BCL. Perinatal correlates of major and minor neurological dysfunction at school age: A multivariate analysis. Develop Med Child Neurol 30:472-481, 1988.
- Klipstein CA, McBride MC. Predictors of cerebral palsy in perinatal hypoxic-ischemic encephalopathy (abstract). Ann Neurol 32:478, 1992.
- Nelson KB, Leviton A. How much of neonatal encephalopathy is due to birth asphyxia? Am J Dis Child 145:1325-1331, 1991.
- Nelson KB, Ellenberg JH. Antecedents of cerebral palsy. I. Univariate analysis of risks. Am J Dis Child 139:1031-1038, 1985.
- Torfs CP, van den Berg BJ, Oechsli FW, Cummins S. Prenatal and perinatal factors in the etiology of cerebral palsy. J Pediatr 116:615-622, 1990.
- Adamson SJ, Alessandri LM, Badawi N, et al. Predictors of neonatal encephalopathy in full-term infants. Brit Med J 311:598-602, 1995.
- Portman RJ, Carter BS, Murphy MG et al. Predicting neonatal morbidity after perinatal asphyxia: A scoring system. Am J Obstet Gynecol 162:174-182, 1990.
- Fee SC, Malee K, Deddish R et al. Severe acidosis and subsedluent neurologic status. Am J Obstet Gynecol 162:802-806, 1990.
- Dennis J, Johnson MA, Mutch LM et al. Acid-base status at birth in term infants and outcome at 4.5 years. Am J Obstet Gynecol 161:213-220, 1989.
- Goldaber KG, Gilstrap LE III, Leveno KJ et al. Pathologic fetal acidemia. Obstet Gynecol 78:1103-1107, 1991.
- Goodwin TM, Belai I, Hernandez P et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol 162:1506-1512, 1992.
- Winkler CL, Hauth JC, Tucker M et al. Neonatal complications at term as related to the degree of umbilical artery acidemia. Am J Obstet Gynecol 164:637-41, 1991.
Assessment of Outcomes Following Neonatal Asphyxia
Jane E. Stewart, M.D., M.S.
Joint Program in Neonatology, Beth Israel, Brigham and Women's and
Children's Hospitals, and Department of Pediatrics, Harvard Medical School
Marie C. McCormick, M.D., Sc.D.
Department of Maternal and Child Health,
Harvard School of Public Health
Alan Leviton, M.D.
Neuroepidemiology Unit, Department of Neurology
Children's Hospital, Boston, Massachusetts
I know that most men (and women)…can seldom discern even the simplest and most obvious truth if it be such as obliges them to admit the falsity of conclusions they have formed,…—conclusions of which they are proud, which they have taught the others, and on which they have built their lives (Tolstoy).
Introduction
Because Bryce and his colleagues1 provided a reasonably complete review of this topic through 1985, we have confined our attention to a review of the literature published since then. Unfortunately, many of the problems identified in that review persist. Thus, we have devoted much of this chapter to the persisting problems.
Problems with Asphyxia Definitions
'Then you should say what you mean,' the March Hare went on. 'I do,' Alice hastily replied, 'at least—at least I mean what I say—that's the same thing, you know.'
'When I use a word,' Humpty Dumpty said in a rather scornful tone, 'it means just what I choose it to mean—neither more nor less (Carroll).'
These citations from Alice's Adventures in Wonderland are quoted because some authors do not write as clearly as their readers would like. Some examples of asphyxia definitions are provided in Table 1. We are especially interested in operational definitions (i.e., those that include criteria that allow a reader to replicate the author's study, should she ever feel so inclined). Unfortunately different authors continue to use the term asphyxia differently2-15 (Table 1).
Stedman's medical dictionary16 defines asphyxia as "1) Unconsciousness due to suffocation or interference of any kind with oxygenation of the blood. 2) Absence of the pulse beat. 3) Cyanosis, local or general, through interference with the circulation." Because we are especially interested in perinatal asphyxia, consider the definition of Snyder and Cloherty,17 "Perinatal asphyxia is an insult to the fetus or the newborn due to lack of oxygen (hypoxia) and/or lack of perfusion (ischemia) to various organs."
Because of the problems posed by multiple different definitions, some investigators have replaced the definition of asphyxia with correlates of asphyxia including:
- Meconium passage in utero;
- Fetal heart (tocographic) monitoring abnormalities;
- Low pH values of scalp or umbilical cord blood;
- Low Apgar scores at either 1 or 5 minutes; and
- Low biophysical profile scores.
Problems with these correlates of fetal distress have been identified by others.18,19 For example, in the Collaborative Perinatal Project, expulsion of meconium during labor occurred in 18 percent of babies.20 Are we to assume that 18 percent of babies are asphyxiated?
TABLE 1: Examples (Published Since 1985) of Operational Definitions of Asphyxia
I THE FIRST DEFINITION REQUIRES THAT MULTIPLE CRITERIA BE MET:
- Required mechanical ventilation and
- Two of the three following criteria:
- Perinatal distress defined by abnormal heart rate patterns or prolonged resuscitative efforts,
- 5-minute Apgar score £ 4, and/or
- Metabolic acidosis defined as a base deficit of > 15 mEq/l in blood obtained during the first hour of life.2
II THE SECOND DEFINITION REQUIRES THAT ALL CRITERIA BE MET:
- Fetal distress,
- Apgar score <6 at 5 minutes,
- Necessity for positive pressure ventilation for >2 minutes, and
- Metabolic acidosis with pH <7.2 within 2 hours of birth.3
III THE THIRD DEFINITION REQUIRES THAT THREE OF THE FOUR CRITERIA BE MET:
- Fetal distress documented on fetal heart rate monitoring (late or variable decelerations),
- Presence of meconium stained amniotic fluid with infant in vertex position,
- Need for endotracheal intubation at delivery, or
- Occurrences of abnormal muscle tone and seizures within 24 hours of age.4
IV THE FOLLOWING DEFINITIONS REQUIRE THAT ONLY ONE OF SEVERAL CRITERIA BE MET IN EACH GROUP:
- Evidence of fetal distress,
Apgar score <6 at 5 minutes,
Positive pressure ventilation for 2 minutes or more, or
Metabolic acidosis (pH £ 7.2 within 2 hours of birth).5 - Fetal bradycardia (heart rate less than 80 beats per minute for at least 60 seconds) or evidence of late decelerations during labor as seen by fetal monitoring,
Apgar score less than 5 at 5 minutes,
Requirement of positive pressure ventilation for at least 2 minutes following delivery, or Acidosis (pH <7.1within the first hour of life).6 - 5-minute Apgar score of 5 or less in infants with or without initial intubation,
5-minute Apgar score of 6 in intubated infants, or
An umbilical cord arterial pH <7.2 or umbilical cord arterial PC02>50 mm Hg.7 - An Apgar score at 1 minute of <7,or
Time to spontaneous respiration of more than 2 minutes.8 - Umbilical arterial blood pH values <7.15, or
Apgar scores of 0-4 at 5 minutes.9 - Apgar score <3 at 1 minute.10
- Evidence of a metabolic acidosis at delivery with umbilical artery buffer base
<34mmol/L11 - Intrapartum fetal distress on fetal heart monitor,
Immediate neonatal distress indicated by a low in 5-minute Apgar (<5), or
Immediate neonatal resuscitation requiring bag and mask or intubation.12
THE FOLLOWING DEFINITIONS ARE NOT OPERATIONAL:
Birth asphyxial factors including:
Abruptio placentae, tight knot in umbilical cord, active labor greater than 20 hours, tumultuous labor, maternal shock, severe fetal hemorrhage, umbilical cord tight around neck or body, umbilical cord prolapse, arrested progress of active labor persistent high uterine tone during labor, maternal anesthesia accident, severe vaginal bleeding at delivery.13
Not defined at all.14
Respiratory problems diagnosed at, or after, birth.15
A committee opinion published by the American College of Obstetricians and Gynecologists included the following: "Intrapartum asphyxia implies fetal hypercarbia and hypoxemia, which, if prolonged, will result in metabolic acidemia. Because the intrapartum disruption of uterine or fetal blood flow is rarely, if ever, absolute, asphyxia is an imprecise, general term. Terms such as hypercarbia, hypoxia, metabolic acidemia, and respiratory or lactic acidemia are more precise.21 Indeed, recently published articles have addressed the neonatal and later characteristics of babies with neonatal acidemia.22-27
Although the level of acidemia required for a definition of asphyxia varies (Table 1), the pH cut off for defining pathologic fetal acidemia appears to be closer to 7.0 than to 7.2.22-25 The value of cardiac dysfunction28 and biophysical profile29,30 are also gaining greater acceptance as indicators/predictors of early neonatal well being.
The limitations of arterial blood gas assessments31 have directed attention away from peripheral blood to measures and correlates of intracranial energy metabolism. This has prompted some to emphasize the brain when seeking early expressions of the presumed adverse developmental effects of asphyxia, including:
- Expressions of damage to brain, especially MRI abnormalities;6,14
- Newborn encephalopathy;19
- Impaired cerebral blood flow regulation;32,33 and
- Near infrared spectrographic documentation of energy metabolism disorders.34,35
Criteria for Outcomes
The criteria for selecting appropriate outcome measures for a given situation generally require that the outcomes be linked to anatomical/physiologic derangement, or be derived from another theoretical framework. The importance of a well specified theoretical model lies not only in the ability to strengthen understanding of the pathogenesis and potential prevention of adverse outcome, but also to anticipate potential confounders, i.e., factors that may contribute to or even account for the observed outcomes other than the event of interest. The lack of a well articulated model incurs the risk of using available, not necessarily appropriate, outcome measures, and failing to ascertain or control for important correlates of outcome.
The criteria for defining asphyxia have already been noted. Most of these criteria can be characterized as clinical markers of generalized physiologic derangement but not specific anatomic damage or alteration in function. Much of the information in potential anatomic and functional impact of this derangement is derived from animal studies and postmortem examination of humans, neither providing a sound basis for predicting specific later morbidity in surviving children. Thus, the selection of outcome measures has been driven largely by what is available. However, even in applying available measures, investigators have often failed to consider the theoretical context of some outcome measures in their interpretation and selection of covariates.
General Selection Criterion: Validity and Reliability
Valid measures are an expression of the concept or construct intended to be measured. An often tacit assumption is that the concept or construct being measured has been adequately specified and the factors influencing the measure are known. For example, not only does the concept of health include indicators of well-being or illness, but also influences on health such as poverty and the consequences of poor health such as death, disability and the need for health services. Establishing validity is complex and may involve examination of the content of measures, comparisons with previously established measures (sensitivity/specificity is a subset of this type of validity testing), and repeated use under a variety of conditions.
Reliability refers to the ability of a given measure to provide the same response on repeated observations. Intrarater reliability assesses the extent to which repeated observations on the same individual by the same observer yield the same response. Interrater reliability reflects the ability of different observers to obtain similar responses.
We have already argued that the concept or construct of neonatal asphyxia still lacks specification and agreement. That, in and of itself, does not preclude selecting outcome measures of potential relevance. In doing so, however, it is essential that attention be paid to the conceptual basis of the outcome measure.
Specific Outcome Measures and Concerns
Mortality. Mortality tends to be validly and reliably measured. Attribution of mortality to asphyxia, however, requires elimination of other causes of death. Obvious major malformations of known natural history represent a straightforward case. Less clear are some cases of immaturity or sepsis.
Specific Diagnoses. The most frequently reported diagnoses associated with asphyxia are cerebral palsy (CP) and seizure disorders. Shaywitz37 has identified many of the issues involved in the diagnosis of CP. The general consensus is that CP is a "nonprogressive disorder of movement and posture due to a defect or lesion of the immature brain" (Bax as quoted by Shaywitz).37 Eliminated are progressive or transient disorders, and CP associated with well defined congenital malformations. However, as Shaywitz notes, little agreement surrounds the age of assessment, classification of types of CP, or severity. Thus, the outcomes being measured may differ substantially across studies.
Shaywitz37 also reports on the interrater reliability of the diagnosis. Among six experienced clinicians the percent agreement was 50 percent with Kappa in the range of 28 percent. Thus, even with agreement in definitions, detection would also vary substantially across observers.
Similar concerns can be raised concerning the diagnosis of seizure disorders. Seizures in the neonatal period are sometimes difficult to diagnose because of their variable appearance and occasional lack of correlation between clinically apparent seizures and electroencephalographic findings.38
Abnormalities on Physical/Neurologic Examination. Other studies have avoided the pitfalls of diagnostic validity by reporting observed abnormalities. In their review, Bryce et al.1 note outcomes such as neurologically abnormal, abnormal tone and abnormal grasp. Such measures incur two difficulties. While not well documented in the literature, they are likely to incur the low interrater reliability associated with most maneuvers on the physical examination.39 In addition, the prognostic significance of isolated abnormal signs or single neurologic examinations is often unclear. Standardized approaches40,41 depend on assessing a number of neurologic functions over time to assess the presence of a neurologic abnormality.
Mental Retardation/Cognitive and Social-Emotional Developmental Delay. In contrast, reliance on cognitive assessments as outcome measures is attractive because of the availability of well standardized assessment instruments with established prognostic significance. Difficulty arises in the specificity of the linkage between the signs of neonatal asphyxia and alterations of development. Several factors may influence causal inferences between perinatal events and later developmental outcomes.
- Developmental abnormalities observed early in infancy may be transient or of limited prognostic significance. Thus, permanent, significant residua are best assessed later in infancy or early childhood.
- The etiology and natural history of variations in developmental patterns are not well established. While overall scores, some major subscales and individual items may have population norms, the significance of more subtle groups of relative strengths and abnormalities is not well established. For example, the most frequently used infant test, the Bayley, has two major components: mental and motor scales. Further refinement into more subscales remains unconventional, and the significance of differential development across these subscales is still unknown.
- Higher order development and its failure, which may result in school difficulties or learning disabilities, should not be assessed until substantially later in childhood. Assessments at later ages, however, may well be highly confounded by intervening events. For example, extensive evidence supports the adverse effects of poverty on cognitive development and maternal mental health on social-emotional development—effects that may far outweigh the influence of all but the most severe perinatal events.42, 43
Disability/Handicap.* Because of the infrequency and variability of abnormalities potentially encountered, investigators have categorized outcomes in terms of functional impact, described as disability or handicap. As has been pointed out in other contexts,44, 45 the definitions of disability/ handicap, and its severity, tend to be idiosyncratic to the individual report. Moreover, such categorizations tend to be composites of performance on standardized tests and/or diagnoses and neurologic abnormalities, thus incurring previously mentioned concerns about reliability and validity.
Moreover, much of the literature on disability/handicap antecedes or lacks modern and standardized approaches. Over the past 10 years, several investigators have modified/extended the types of measures of limitations of activities of daily living (ADLs) suitable for adults to be appropriate for children.46-48 Such approaches are derived from extensive experience46 or comprehensive theoretical models of health status.47, 48 More recently, the Institute of Medicine examined the issue of disability, laying out both a conceptual approach and specific definitions.49 At least one group has operationalized this approach in their examination of outcomes of one neonatal problem, very low birth weight.50
Assessment Criteria
For purposes of this report, the outcomes in the studies reviewed were assessed for the presence of the following criteria.
- Clear and specific definition of the population studied;
- Clear and specific definition of the outcome measure(s);
- Specification of assessment techniques (e.g., type of assessor, training and experience, reliability checks if appropriate, timing);
- Comparability of outcomes with a) nature of neonatal insult (i.e., causal influence) and b) later outcomes if appropriate (i.e., predictive value);
- Appropriate attention to the underlying theoretical model of the outcome measure with identification and ascertainment of confounders; and
- Explicit recognition of the statistical properties of the outcome measures and their effect on statistical power.
Despite the fact that we prefer using the term neonatal encephalopathy or neonatal depression rather than hypoxic-ischemic encephalopathy or asphyxia, we have used the original terminology used by the authors in each of the reports reviewed.
Outcome Studies (Table 2). The most extensive studies that have been published recently are by Robertson and Finer.12, 51-53They include follow-up of a group of 167 term infants born between 1974 and 1979 with evaluations performed at 6, 12 and 27 months, 3.5, 5.5 and 8 years. The criteria used for entry into the study were a diagnosis of hypoxic-ischemic encephalopathy (HIE) defined as an abnormal neurological examination after 1 hour of age and at least one of the following: intrapartum fetal distress, based on abnormal heart-rate patterns; immediate neonatal distress indicated by a low 1- or 5-minute Apgar score (<5); and/or immediate neonatal resuscitation, including bag and mask ventilation or intubation with ventilation. The neurological exam was abnormal if an alteration in consciousness, muscle tone or abnormal primitive reflexes were noted. The HIE was classified into one of Sarnat's three stages: stage I or mild—hyperalertness, hyperexcitability; stage 2 or moderate—lethargy, hypotonia, suppressed primitive reflexes; and stage 3or severe—stupor, flaccidity, absent primitive reflexes.
TABLE 2: Summary of Outcome Studies of Infants with Diagnosis of Perinatal Asphyxia
| AUTHOR | NUMBER OF PATIENTS | ENCEPHALOPATHY CLASSIFICATION | RESULTS | OTHER PREDICTORS OF OUTCOME |
|---|---|---|---|---|
| Robertson (1985, 1988, 1989) | 226 | Yes | 11% Mortality 19% Handicap 10% CP 11% Cognitive delay 5.5% Visual impairment 6.5% Convulsive disorder |
Abnormal neurological exam at discharge |
| Adsett (1985) | 56 | No | 14% Mortality 23% Normal 8% Mild handicaps 42% Major handicaps 27% Indefinite |
CT Scan |
| Levene (1986) | 122 | No | 11% Mortality 8% Severe neurologic abnormality |
Apgar Scores |
| Ishikawa (1987) | 86 | No | 15% Mortality 76% Normal 9% Mild abnormalities 15% Major abnormalities |
Abnormal neurological exam at discharge Persistent abnormal neurological findings Neonatal seizures |
| Low (1988) | 37 | Yes | 50% Normal 27% Minor abnormalities 13% Major abnormalities |
Degree Metabolic acidosis |
| Perlman (1988) | 36 | No | 6% Mortality 17% Abnormal outcome |
Renal Injury |
| Shankaran (1991) | 28 | Yes | 14% Mortality 42% Spastic quadriplegia or hemiplegia 50% Microcephaly & seizure disorder 25% Vision impairment 17% Hearing 54% Language delay 50% Normal cognitive development 38% Severe cognitive delay |
Cardiac injury Renal injury Pulmonary injury Hematologic injury Seizures, EEG, CT |
| Lam (1992) | 40 | Yes | 10% Mortality 7.5% Normal outcome 10% Handicap 2.5% Moderate handicap 2.5% Severe handicap |
Renal injury |
Outcome measures evaluated included the following handicaps: cerebral palsy, cognitive delay, visual impairment, epilepsy, and neurosensory hearing loss > 70 dB. IQ was measured by the Stanford-Binet Intelligence Scale. Visual motor integration, speech and language development, fine and gross motor skills were also assessed as were school readiness and language development at age 5.5 years. At 8 years, a full battery of school performance measures were tested.
IQ tests were administered by a psychologist, speech and language by a speech pathologist, hearing by an audiologist, motor assessments by a physical therapist and developmental pediatrician, and educational testing by a reliability-tested educator; testers were unaware of the patients' neonatal courses. Comparable groups of children with normal healthy neonatal courses were also studied at 5.5 and 8 years. Potential confounding factors such as socioeconomic status, mother's language and intervening illnesses were evaluated.
Eleven percent of the 226 children died and of the 200 survivors, 19 percent had handicap: 11 percent with severe cognitive delay, 10 percent with cerebral palsy, 6.5 percent with convulsive disorders, 5.5 percent with visual loss and 2 percent with deafness. Outcome was significantly related to severity of encephalopathy with all of the severe encephalopathy group developing severe disability, none of the mild encephalopathy children developing disability and 21 percent of the moderate encephalopathy infants developing later disability. In those with moderate encephalopathy, an abnormal neurological exam at the time of discharge and the occurrence of seizures in the neonatal period appeared to be related to worsened outcome. At the 5.5 and 8 year assessments, children who had mild encephalopathy performed as well as their control peers. This led the authors to question whether these infants should be classified to have had encephalopathy at all. Children with moderate and severe encephalopathy however, did less well than their peers on assessments of school readiness and later school performance.51-53
Adsett et al.3used criteria for perinatal asphyxia including: fetal distress (not further defined), Apgar score <6 at 5 minutes, necessity for positive pressure ventilation for >2 minutes, and metabolic acidosis with pH <7.2 in the first 2 hours of life. Fifty-six full-term infants met these criteria, 48 of whom survived and were evaluated by a neurologist or pediatrician at ages 4-23 months with outcomes reported as: Normal—normal neurological examination and development at one year or older; Mild handicap—mild delay in development without definite neurological deficits, or equivocal neurological abnormalities resulting in minor impairment of function; and Major handicap—definite permanent neurological deficits. Only 23 percent were reported as normal, 8 percent as mild handicap, 42 percent as major handicap, and 27 percent as indefinite. CT scans performed on all patients were found to add to prediction of outcome with normal and patchy (areas of hypodensity) abnormalities correlating with better outcomes and diffuse and global (extensive areas of deceased density) abnormalities correlating with poor outcomes. Assessment techniques used were not reported nor was the evaluation of potential confounding factors such as socioeconomic status or intervening illness.
Levene's group54evaluated 122 patients with perinatal asphyxia (definition not provided) born between 1980-1984 and assessed between 1-5 years (median 2.5 years). As in Robertson's studies, patients were categorized as having mild (minor disturbance of tone, hyperalertness, and slight feeding difficulties, recovering by 48 hours after birth); moderate (lethargy, more pronounced abnormalities of tone, poor feeding, and convulsions, with signs of recovery by 7 days); and severe (coma, failure to maintain adequate ventilation, profound hypotonia, and seizures). Children were evaluated by a neurologist and a psychologist (Bayley Scales of Infant Development) if abnormalities were suspected. Outcome was defined as presence or absence of severe neurological abnormality (cerebral palsy sufficient to impair independent locomotion, developmental delay severe enough to warrant special education, sensorineural hearing loss, visual impairment, or epileptic seizures requiring medication) or death. Fourteen patients died. Of the 108 survivors, 8 percent developed severe neurological abnormalities; all with severe handicap had moderate or severe perinatal encephalopathy. Apgar scores were not found to be good predictors of outcome. The evaluation of potential confounders was not described.
Ishikawa et al.55evaluated 86 full-term infants in whom asphyxia was defined by the following criteria: 1-minute Apgar score of <6; neurological complications including: (a) findings of stupor or lethargy, hypotonia, weakness of proximal limbs, abnormal breathing patterns, and jitteriness or seizures, and (b) one or more abnormal findings on laboratory test such as cerebrospinal fluid, cranial ultrasound, cerebral angiography electroencephalogram, or brain CT scan. Children were evaluated from ages 3-13 years (mean 8 yrs, 5 mos) using outcome measures including neurological exam, hearing assessment, and intelligence quotient (IQ) as measured by a clinical psychologist (Wechsler or Tanaka-Binet intelligence tests).
Of the 55 patients followed (75 percent), 15 percent developed major developmental abnormalities (cerebral palsy, mental retardation or epilepsy); and 9 percent developed mild abnormalities (normal function in daily life). Neonatal factors that were found to be of predictive value included: an abnormal neurological exam on discharge; an absent Moro reflex for >6 days; disturbance of consciousness over 6 days; and the occurrence of neonatal seizures and poor sucking over 28 days. No evaluation of potential confounders was noted.
In a report of 37 term infants with neonatal asphyxia, Low et aI.11 used criteria for neonatal asphyxia that included evidence of a metabolic acidosis at delivery (umbilical artery buffer base <34 mmol/L). As Robertson and Levene did, infants were divided into three groups by severity: mild (hyperalertness, irritability, jitteriness, transient hypertonia or hypotonia); moderate (lethargy, severe hypotonia, occasional seizures); and severe (coma, multiple seizures, and recurrent apnea). Patients were evaluated at 6 and 12 months of age with a neurological examination, the Bayley Scales of Infant Development, and the Uzgiris and Hunt scale.
A control group of 76 healthy infants with no history of neonatal complications was also assessed. Twenty-seven percent of the infants with asphyxia developed minor deficits and 13 percent major deficits; 60 percent were reported as normal compared to 92 percent normal in the control group. Assessments of potential confounders such as maternal education or socioeconomic status were not reported.
Perlman and Tack7assessed renal injury in another follow-up evaluation of infants with neonatal asphyxia. Asphyxia criteria included one or more of the following: 1) 5-minute Apgar score <5 with or without need for intubation; 2) 5-minute Apgar 6 if required intubation; and/or 3) umbilical cord arterial pH <7.20 or umbilical cord arterial PCO2 > 50mm Hg. Thirty-six full-term infants born between 1985-1986 were assessed at age 12-18 months with methods that were not reported. Of the six patients with persistent oliguria, only one had a normal outcome; the rest had cerebral palsy and seizures. Transient oliguria and normal urine output were associated with a much better outcome. Overall 17 percent of patients had abnormal outcomes and 5 percent died. Little further information about these infants including potential confounders was provided.
Shankaran et al.4 thoroughly evaluated a small group of 24 survivors with perinatal asphyxia as defined by satisfying at least three of the following four criteria: fetal distress documented on fetal heart monitor (late or variable decelerations); presence of meconium stained amniotic fluid with the neonate in a vertex position; the need for endotracheal intubation at delivery; and the occurrence of abnormal muscle tone and seizures within 24 hours of age. Infants were all full-term and delivered between 1980-1982. Sarnat stages of encephalopathy were assigned to each patient. Patients were evaluated at 3, 6, and 12 months, and then yearly up to 5 years of age with neurological exams and age appropriate standardized developmental assessments (Bayley Scales of Infant Development and McCarthy Scales of Children's Abilities), language and hearing assessments. Information regarding socioeconomic status was routinely collected. During the neonatal time period, information regarding multisystem function including cardiac, renal, pulmonary, hematologic and neurological systems was also evaluated. At 5 years of age, 58 percent of infants had normal motor exams; and the remaining 42 percent had spastic quadriplegia or hemiplegia. Fifty percent had microcephaly and seizure disorders; 25 percent vision impairment; 17 percent hearing and 54 percent language delay; 38 percent had normal cognitive development while 50 percent had very abnormal cognitive development. Factors that were found in multiple regression analysis to be predictive of lower cognitive scores were: lower socioeconomic status, neonatal seizures, renal problems, cardiac problems, need for cardiopulmonary resuscitation, and poor head growth at 3 months of age. Potential confounders including socioeconomic status and intervening illness (meningitis, postnatal brain injury) were also evaluated.
The last report to be reviewed is that of Lam et al.10 describing the outcome of 40 full-term newborns delivered in 1985-1987 who met the single criterion for asphyxia of having an Apgar score of <3 at 1 minute. These infants were classified using Amiel-Tison's staging: (Stage Ia) hyperexcitability and mild abnormalities of tone for £ 7 days, (Ib) for >7 days; (Stage IIa) CNS depression such as lethargy or light coma without seizures, (IIb) with seizures; (Stage III) Deep coma and repetitive seizures without brainstem signs and the presence of spontaneous respiratory efforts, (IIIb) with brainstem signs and the absence of spontaneous respiration efforts. Outcome assessment measures included a full neurodevelopmental exam performed by a neurologist at 2 years of age. Outcome was reported as: Normal—within the average age development and no evidence of handicap; Mild handicap—variations from normal on neurologic or developmental examination without a specific diagnosis; Moderate handicap—trainable retardation, severe behavioral disorders and/or convulsive disorders, mild or moderate neurosensory deafness, spastic diplegia, hemiplegia, visual impairment; Severe handicap—spastic quadriplegia, severe psychomotor retardation, severe neurosensory deafness or blindness; and Death. Fully 75 percent had a normal outcome, 10 percent had mild handicap, 5 percent had moderate or severe handicap and 10 percent died. In this study again outcome was related to the severity of the initial neonatal classification. All children who had stage III encephalopathy developed moderate to severe handicap or death and none of the patients with stage I or II developed later significant sequelae. Severe renal impairment was also again a predictor of poor outcome. Potential confounders were not evaluated.
Conclusions
In summary, studies of the outcome of infants with perinatal asphyxia remain cumbersome to compare because of differences in the basic definitions of asphyxia essential to defining the population of infants being studied, differences in measuring outcomes (Table 3) and in evaluating potential confounding factors that also contribute to those outcomes. Trends in the past 8 years, however, appear to clarify that using only simple criteria such as low Apgar scores or blood pH to define the population at risk for adverse outcome is not sufficient. Classifying the severity of neonatal encephalopathy as five of the eight studies reviewed in this paper did, appears to augment the ability to predict outcome. The diagnosis of mild encephalopathy, on the other hand, appears to be unnecessary as the outcome of these patients appears to be universally good. The majority of these studies also attempted to use evidence of actual end-organ insult as demonstrated by persistent abnormal neonatal neurological state, seizures or evidence of injury to other organ systems such as the kidneys or heart and found this to provide better prediction of risk for an adverse outcome.
TABLE 3: Design of Outcome Studies of Infants with Diagnosis of Perianatal Asphyxia
| AUTHOR | STANDARDIZED OUTCOME MEASURES | CONTROL GROUP | ASSESSMENT OF POTENTIAL CONFOUNDERS |
|---|---|---|---|
| Robertson | Yes | Yes | Yes |
| Adsett | No | No | No |
| Levene | Yes | No | No |
| Ishikawa | Yes | No | No |
| Low | Yes | Yes | No |
| Perlman | No | No | No |
| Shankaran | Yes | No | No |
| Lam | No | No | No |
Perhaps with new, more precise methods of assessing CNS injury and clear definitions of neonatal encephalopathy with criteria that are universally applied, the group at greatest risk of poor outcome will also be better identified so that interventions can be offered as early as possible to optimize potential development. Likewise, in the continuing follow-up of these infants, standard methods of assessment, use of comparison groups and evaluation of factors such as socioeconomic status and parental education should routinely be utilized in the analysis of outcome results. Until these definitions and methods of assessment are standardized, studies can not be compared and results can not be generalized to the general population of infants.
References
- Bryce RL, Halperin ME, Sinclair JC. Association between indicators of perinatal asphyxia and adverse outcome in the term infant: A methodological review. Neuroepidemiol 4:24-38, 1985.
- Goldberg RN, Moscoso P, Bauer CR et al. Use of barbiturate therapy in severe perinatal asphyxia: A randomized controlled trial. J Pediatr 109:851-856, 1986.
- Adsett DB, Ritz CR, Hill A. Hypoxic-ischaemic cerebral injury in the term newborn: Correlation of CT findings with neurological outcome. Dev Med Child Neurol 27:155-160, 1985.
- Shankaran S, Woldt E, Koepke T et al. Acute neonatal morbidity and long-term central nervous system sequelae of perinatal asphyxia in term infants. Early Hum Dev 25:135-148, 1991.
- Roland EH, Jan JE, Hill A, Wong PK. Cortical visual impairment following birth asphyxia. Pediatr Neurol 2:133-137, 1986.
- Lupton BA, Hill A, Roland EH et al. Brain swelling in the asphyxiated term newborn: Pathogenesis and outcome. Pediatrics 82:139-146, 1988.
- Perlman JM, Tack ED. Renal injury in the asphyxiated newborn infant: Relationship to neurologic outcome. J Pediatr 113:875-879, 1988.
- Blair E, Stanley FJ. Intrapartum asphyxia: A rare cause of cerebral palsy. J Pediatr 112:515-519, 1988.
- Riikonen RS, Kero PO, Simell OG. Excitatory amino acids in cerebrospinal fluid in neonatal asphyxia. Pediatr Neurol 8:37-40, 1992.
- Lam BCC, Yeung CY. Perinatal features of birth asphyxia and neurologic outcome. Acta Paediatr JPN 34:17-22, 1992.
- Low JA, Galbraith RS, Muir DW et al. Motor and cognitive deficits after intrapartum asphyxia in the mature fetus. Am J Obstet Gynecol 158:356-361, 1988.
- Robertson C, Finer N. Term infants with hypoxic-ischemic encephalopathy: Outcome at 3.5 years. Dev Med Child Neurol 27:473-484, 1985.
- Naeye RL, Peters EC, Bartholomew M, Landis JR. Origins of cerebral palsy. Am J Dis Child 143:1154-1161, 1989.
- Barkovich AJ, Truwit CL. Brain damage from perinatal asphyxia: Correlation of MR findings with gestational age. AJNR 11:1087-1096, 1990.
- Foley J. Dyskinetic and dystonic cerebral palsy and birth. Acta Paediatr 81:57-60, 1992.
- Taylor NB (ed). Stedman's Medical Dictionary. Baltimore: The Williams and Wilkins Company, 19th Revised Edition, pp 137, 1957.
- Snyder EY, Cloherty JP. Perinatal asphyxia. In: Cloherty JP, Stark AR (eds). Manual of Neonatal Care. Boston: Little Brown and Company, pp 393-419, 1991.
- Hollander DI, Wright L, Nagey DA et al. Indicators of perinatal asphyxia. Am J Obstet Gynecol 157:839-43, 1987.
- Nelson KB, Leviton A. How much of neonatal encephalopathy is due to birth asphyxia? AJDC 145:1325-1331, 1991.
- Nelson KB, Ellenberg JH. Obstetric complications as risk factors for cerebral or seizure disorders. JAMA 251:1843-1848, 1984.
- Committee on Obstetrics: Maternal and Fetal Medicine. Utility of umbilical cord blood acid-base assessment. American College of Obstetricians & Gynecologists Committee Opinion Number 91, 1991.
- Fee SC, Malee K, Deddish R et al. Severe acidosis and subsequent neurologic status. Am J Obstet Gynecol 162:802-806, 1990.
- Low JA, Muir DW, Pater EA, Karchmar EJ. The association of intrapartum asphyxia in the mature fetus with newborn behavior. Am J Obstet Gynecol 163:1131-1135, 1990.
- Goldaber KG, Gilstrap LC 3rd, Leveno KJ et al. Pathologic fetal acidemia. Obstet Gynecol 78:1103-1107, 1991.
- Mires GJ, Agustsson P Forsyth JS, Patel NB. Cerebral pathology in the very low birthweight infant: Predictive value of peripartum metabolic acidosis. Eur J Obstet Gynecol Reprod Biol 42: 181-185, 1991.
- Winkler CL, Hauth JC, Tucker JM et al. Neonatal complications at term as related to the degree of umbilical artery acidemia. Am J Obstet Gynecol 164:637-641, 1991.
- Goodwin TM, Belai I, Hernandez P et al. Asphyxial complications in the term newborn with severe umbilical acidemia. Am J Obstet Gynecol 167:1506-1512, 1992.
- Gill AB, Weindling AM. Echocardiographic assessment of cardiac function in shocked very low birthweight infants. Arch Dis Child 68:17-21, 1993.
- Vintzileos AM, Campbell WA, Rodis JF et al. The relationship between fetal biophysical assessment, umbilical artery velocimetry, and fetal acidosis. Obstet Gynecol 77:622-626, 1991.
- Walkinshaw S, Cameron H, MacPhail S, Robson S. The prediction of fetal compromise and acidosis by biophysical profile scoring in the small for gestational age fetus. J Perinat Med 20:227-232, 1992.
- Dudell G, Cornish JD, Bartlett RH. What constitutes adequate oxygenation? Pediatrics 85:39-41, 1990.
- Pryds O. Control of cerebral circulation in the high-risk neonate. Ann Neurol 30:321-329, 1991.
- Wyatt JS, Edwards AD, Cope M et al. Response of cerebral blood volume to changes in arterial carbon dioxide tension in preterm and term infants. Pediatr Res 29:553-557, 1991.
- Bucher H-U, Edwards AD, Lipp AE, Duc G. Comparison between near infrared spectroscopy and133xenon clearance for estimation of cerebral blood flow in critically ill preterm infants. Pediatr Res 33:56-60, 1993.
- Hirtz DG. Report of the National Institute of Neurological Disorders and Stroke workshop on near infrared spectroscopy. Pediatrics 91:414 -417, 1993.
- Carmines EG, Zeller RH. Reliability and validity assessment. Quantitative Applications in the Social Sciences Series, #17, Beverly Hills: Sage Publications, 1979.
- Shaywitz BA. The sequelae of hypoxic-ischemic encephalopathy. Sem Perinatol 11:180-190, 1987.
- Mizrahi EM. Neonatal seizures: Problems in diagnosis and classification. Epilepsia 28:546-555, 1987.
- Feinstein AR. A bibliography of publications on observer variability. J Chron Dis 38:619-32, 1985.
- Amiel-Tison C, Grenier A (eds). Neurological Assessment During the First Year of Life. New York: Oxford University Press, 1986.
- Swanson MW, Bennett FC, Shy KK, Whitfield ME. Identification of neurodevelopmental abnormality at four and eight months by the movement assessment of infants. Dev Med Child Neurol 34:321-337, 1992.
- McCormick MC, Brooks-Gunn J, Workman-Daniels K et al. The health and developmental status of very low birth weight children at school age. J Am Med Assoc 267:2204-2208, 1992.
- McCormick MC, Workman-Daniels K, Brooks-Gunn J. The behavioral and emotional well-being of school-age children with different birthweights. Pediatrics (in press).
- Kitchen WH, Ryan MM, Rickards A et al. Changing outcome over 13 years in very low birth weight infants. Seminars in Perinatol 6:373-389, 1982.
- Escobar GJ, Littenberg B, Petitti DB. Outcome among surviving very low birthweight infants: A meta-analysis. Arch Dis Child 66:204-211, 1991.
- Stein REK, Jessop DJ. Functional Status II(R). A measure of child health status. Med Care 28:1041-1055, 1990.
- Eisen M, Donald CA, Ware JE et al. Conceptualization and Measurement of Health of Children in the Health Insurance Study. R-2313-HEW Santa Monica: The Rand Corporation, 1980.
- Kaplan RM, Anderson JP. A general health policy model: Update and applications. Health Serv Res 23:203-235, 1988.
- Institute of Medicine. Disability in America. Washington, DC: National Academy Press, 1991.
- Schreuder AM, Veen S, Ens-Dokkum MH et al. Standardised method of follow-up assessment of preterm infants at the age of 5 years: Use of the WHO classification of impairments, disabilities and handicaps report from the collaborative project on preterm and small for gestational age infants (POPS) in The Netherlands, 1983. Paediatr and Perinatal Epidemiol 6:363-380, 1992.
- Robertson CMT, Finer NN. Educational readiness of survivors of neonatal encephalopathy associated with birth asphyxia at term. J Dev Behav Pediatr 9:298-306, 1988.
- Robertson CMT, Finer NN, Grace MGA. School performance of survivors of neonatal encephalopathy associated with birth asphyxia at term. J Pediatr 114:753-60, 1989.
- Robertson CMT, Finer NN. Long-term follow-up of term neonates with perinatal asphyxia. Clin Perinatol 20:483-499, 1993.
- Levene MI, Sands C, Grindulis H, Moore JR. Comparison of two methods of predicting outcome in perinatal asphyxia. Lancet: 67-69, 1986 (Jan).
- Ishikawa T, Ogawa Y, Kanayama M, Wada Y. Long-term prognosis of asphyxiated full-term neonates with CNS complications. Brain Dev 9:48-53, 1987.
Discussion
Alan Leviton, M.D.
Children's Hospital, Boston, Massachusetts
These two papers are clear, reasonably complete and cogent. Thus, I need not address their inadequacies. Rather, I would prefer to unify them where possible and then suggest a menu of options to help rectify the deficiencies of the literature.
Let me begin by pointing out that I'm not even sure we could all agree on a definition of asphyxia. For purposes of this discussion I will use the term acute perinatal asphyxia as metabolic disturbances resulting in the loss of cellular integrity within the brain. I offer the provocative view that we need to consider glia as well as neurons.
I'm not sure what the time bounds should be for perinatal. In part my uncertainty/confusion reflects Karin Nelson's point that disturbances during the birth process might have their origin months before. First suggested by Freud 100 years ago, this view has been bolstered by some recent studies identified by Dr. Nelson.
Both papers emphasize the heterogeneity of acute perinatal asphyxia. I'd like to expand on this. Some babies with newborn encephalopathy were born during or after an obstetrical/maternal catastrophe (Table 1, Model 1). This model, the acute perinatal asphyxia leads to newborn encephalopathy model, is the focus of this meeting. Leads to here is short for increases the probability of. Karin Nelson points out that we have two other models. In the Freudian model early pregnancy disturbances lead to acute perinatal asphyxia, which in turn leads to newbom encephalopathy (Model 2). In a third model, the early pregnancy disturbances themselves damage the brain (Model 3). This model need not invoke newborn encephalopathy. (Models without newborn encephalopathy are not presented.) When newborn encephalopathy occurs in Model 3 it can be viewed as a marker of pre-existing damage and an expression of impaired adaptation from a liquid to a gas environment.
In these three models newborn encephalopathy represents brainstem dysfunction (i.e., impaired suck, swallow, respiration regulation and consciousness). Improvement allows survival.
Those babies with involvement of the brain beyond the brainstem are the ones most at risk of later handicap. Thus, each of the three models has some babies who will recover substantially, and others who will later manifest significant neurologic impairment.
As I see it, one of the goals of this meeting is to explore ways that might help discriminate those newborn encephalopathy babies who are most likely to improve from those who are least likely.
What follows is my menu of needed studies.
Advances in Measuring Antecedents, Acute Dysfunction, and Later Function
We need to be able to identify metabolic or newborn encephalopathy characteristics that predict later dysfunction. First I believe we need a better understanding of what is needed to preserve cellular integrity. Improvements in measuring intracranial energy metabolism in the fetus and newbom seem to be worthy targets of study. So, too, are techniques that would allow measurement of phenomena that stave off apoptosis (i.e, programmed death).
I'm not a basic scientist, so I don't feel I can be more specific. I do know, however, that many of the epidemiologic advances in identifying the antecedents and correlates of perinatal and neonatal disturbances have, and will be, based on technologic advances.
We also need to find answers to such questions as: what are the limitations to these technologic advances? How soon after an acute metabolic disturbance will available measurements return to normal? Thus I ask NIH to support basic research that we epidemiologists can build on.
Advances in assessing placental function and fetal well-being hold the promise of providing some information about some of the pre-labor disturbances that might lead to acute perinatal asphyxia. I make this point to counterbalance the emphasis of other speakers on intrapartum events.
TABLE 1: Six Models Intended to Demonstrate the Heterogeneity of Newborn Encephalopathy
Abbreviations:
APA - Acute Perinatal Asphyxia
NE - Newborn Encephalopathy
(+) - Present
(-) - Absent
(X) - Pregnancy perturbation leading to or associated with APA
(Y) - Pregnancy perturbation leading to disturbance of normal brain development
| MODEL | TRIMESTER First Second Third |
LABOR AND DELIVERY | FIRST POSTNATAL DAYS | DISABILITY YEARS LATER |
|---|---|---|---|---|
| A 1 B A |
|
APA APA APA |
NE NE NE |
+ - + |
| 2 B A |
X Y |
APA | NE NE |
+ - |
| 3 B |
Y |
NE |
- |
Dr. Stewart and her colleagues emphasize the need for improved, standardized methods of assessing later function and of dealing with potentially confounding intervening events that might obscure the relationship between acute perinatal asphyxia/newborn encephalopathy and later dysfunction. These are areas worthy of additional study and funding.
Studies of Phenomena that Contribute to Newborn Encephalopathy
Some babies with newborn encephalopathy were born during or after an obstetrical or maternal catastrophe (Models 1 and 2). This does not mean that overwhelming placental dysfunction accounts for most or all newborn encephalopathy. What proportion of babies with newborn encephalopathy experience apparently uneventful labor and delivery? In essence, how much of all newborn encephalopathy is represented by Model 3? This type of information is very important if we are to deal with preventing intrapartum disturbances that lead to newborn encephalopathy. We need to be able to identify and separate the later pregnancy causes of newborn encephalopathy from earlier pregnancy perturbations that damage the brain and impair the newborn's adaptation from an amniotic fluid to an air environment. Wouldn't it be better to prevent newborn encephalopathy than to treat it?
This type of information, however, is also important for clinical trials of what Karin Nelson calls damage control. A potentially toxic drug should not be administered needlessly to those babies whose newborn encephalopathy had its origin weeks to months before delivery.
Both papers emphasize the limitations of what is known. I compliment Dr. Nelson and my coauthors and underscore their plea for more work in these areas.
Discussion
James A. Lemons, M.D.
Section of Neonatal-Perinatal Medicine,
Indiana University Medical Center, Indianapolis
Clinical Studies of Long-term Outcome
The elegant discussions and reviews by Drs. Nelson, Stewart, McCormick and Leviton raise a number of important issues warranting further emphasis. Of primary importance is the fact that perinatal asphyxia in the term infant is an uncommon event with rare adverse outcome. We lack a specific definition of the term asphyxia and are generally unable to ascertain with any degree of certainty the duration, frequency, number, severity or timing of the insults in relation to birth. It is no wonder that studies attempting to delineate predisposing factors, biochemical/physiologic/radiologic correlates or adverse outcomes have yielded mixed and often conflicting results.
As obstetric and neonatal care have improved dramatically over the past 20 years, the incidence of avoidable perinatal injury to the term fetus has declined, perhaps to a level that may be difficult to reduce further. Nonetheless, at a time when chances for delivery of a healthy term infant have continued to improve, litigation has risen seemingly exponentially. Pressures have come to bear on the medical community, particularly obstetricians, to accurately define perinatal asphyxia and to document how asphyxia may be related to long-term neurologic sequelae. There is now compelling evidence that birth asphyxia leads to adverse neurologic outcome or cerebral palsy in a small minority of instances, and conversely the majority of cases of cerebral palsy cannot be attributed to perinatal asphyxia. Some have interpreted this lack of one-to-one cause and effect relationship to indicate that asphyxia rarely if ever, causes permanent central nervous system (CNS) injury in and of itself. In other words there is always an underlying abnormality of the fetus, placenta, or mother which has already caused significant injury or predisposes the infant to potential damage from an asphyxic insult. This view does not seem tenable.
Based upon the data available to date and as reviewed by Nelson, Stewart et al. asphyxia clearly does produce permanent and sometimes severe brain injury. It is also true (as for all biology) that we may anticipate a wide spectrum in the vulnerability of the fetus to an insult, the physiologic response of the fetus/placenta/mother to that insult and the recoverability of the fetus from that insult. This would be true even if the asphyxial insult were uniform in its severity duration, and timing—which we recognize is hardly ever the case in the clinical setting. Even animal studies suggest there is great diversity in the fetal response to a standard asphyxial insult.
Based upon reported studies, it is clear that Apgar scores and early pH measurements are not by themselves predictive of either short or long-term neurologic morbidity, except at extreme values. The strongest correlate with long-term neurologic outcome is short-term neurologic outcome. Moderate or severe neonatal encephalopathy as characterized clinically by Sarnat and Sarnat is relatively highly predictive of abnormal long-term outcome. Unfortunately, the manifestations of moderate to severe neonatal encephalopathy generally do not become apparent for several hours or days after birth. Such a delay in onset of clinical symptomatology may preclude timely diagnosis necessary for effective interventions, if we have no other methods to confirm the insult earlier in the hospital course. It may well be that the CNS damage related to the asphyxial insult may be a continuum and possibly related to other physiologic and other biochemical changes occurring during and shortly after the insult. For instance, myocardlial insufficiency secondary to an hypoxic-hypercarbic insult may in some instances cause severe compromise in cerebral profusion during the first minutes or hours after birth. By the time hypoxic-ischemic encephalopathy is manifest clinically, cardiac function may have returned to normal. At this time we appear to lack objective and highly specific markers which may be used to identify either the fetus or neonate in the first minutes after birth who is likely to develop moderate or severe hypoxic-ischemic encephalopathy. Validation of a scoring system such as proposed by Portman et al. may provide us with a valuable clinical tool, which would identify such infants by one hour of age. Unless such tools/markers become available, epidemiologic studies of perinatal asphyxia must include large numbers of infants who are going to be normal in order to study relatively small numbers of infants who will have significant long-term sequelae.
In almost all cases, we are primarily interested in neurologic (rather than other organ) morbidity as it relates to an asphyxial insult. Generally, other organ systems which experience hypoxic-ischemic damage recover fully in newborn infants. In those rare instances when this is not true, the brain injury is generally overwhelming. Therefore, it is reasonable that the clinical definition of perinatal asphyxia be based primarily upon neonatal encephalopathic changes. Recognizing that the causes of the asphyxic insult in terms of altered profusion, oxygenation, and ventilation may vary considerably in terms of severity, duration, and timing, we need to seek out specific markers which will predict HIE. Such markers need to be accessible and measurable before or shortly after birth. Only then will we be able to ascertain whether other predisposing factors existed within the fetus, whether other mediators of injury such as cytokins were present, and whether proposed interventions are effective and safe. As Dr. Nelson indicates, new findings in cell biology and developmental neurobiology are likely to shed additional light on defining the vulnerability of the immature brain to possible hypoxic-ischemic insult. Randomized clinical trials (using sound epidemiologic principles) must be undertaken with great care, based upon solid scientific evidence accrued from in vitro investigation, animals models and other valid clinical studies.
From a practical point of view, early diagnosis of devastating neurologic injury is important in guiding clinical decision making by the care takers and the infant's parents/guardians. If effective treatment is not available, then decisions regarding continuation or withdrawal of life support may be appropriate and necessary. Such decisions must be made early in the clinical course of the infant, as in most instances the life threatening manifestations of brain injury (i.e., effect on respiratory drive) tend to abate after the first 48-72 hours.
Whether asphyxia is a term which we should continue to use may be debated. If it is to be used, a clear definition from both a clinical and pathophysiologic perspective is imperative. Currently asphyxia is defined primarily upon outcome rather than any objective characterization of the insult itself. Until we can qualitatively and quantitatively measure the asphyxial insult and the vulnerability of the fetal/placental/maternal unit to the insult, we may not be able to further reduce morbidity from perinatal asphyxia.
Session VI: Clinical Research
Moderator: Jerold F. Lucey
Methodologies for Documenting Timing and Evidence of Brain Asphyxia Including NIR and NMR
A.D. Edwards, M.D.
Department of Paediatrics and Neonatal Medicine,
Royal Postgraduate Medical School, London
Introduction
The development of non-invasive methods for investigating the structure and function of the brain in small and ill infants allows clearly defined questions to be addressed about potentially brain damaging lesions, particularly those arising in the perinatal period. This report will describe three of the available methods for non-invasive investigation of birth asphyxia; magnetic resonance spectroscopy, near infrared spectroscopy and cerebral electrophysiological measurements.
Magnetic Resonance Spectroscopy
Phosphorus Magnetic Resonance Spectroscopy31
P magnetic resonance spectroscopy enables direct observations to be made of phosphocreatine (PCr) and inorganic phosphate (Pi) as well as ATP. In situations where the processes of oxidative phosphorylation are impaired either because the oxygen supply to the tissue is curtailed or the mitochondrial mechanisms for consuming oxygen are damaged, the concentration of ATP will tend to fall. Initially the fall is extremely small because of the buffering effect of the creatine kinase reaction, which maintains the ATP concentration almost constant, but at the same time causes a reduction in PCr concentration and an almost reciprocal rise in that of Pi. The concentration ratio PCr/Pi (which is related to the phosphorylation potential) can therefore be used as an index of the extent to which oxidative phosphorylation is impaired. Only when PCr/Pi has fallen to a very low level does the ATP concentration fall appreciably.
Spectra from infants with hypoxic-ischemic brain injury regularly show a reduction on PCr and an increase in Pi.1 As well as a large reduction in PCr/Pi, the concentration of ATP is below normal. Spectra from infants with other forms of hypoxic-ischemic injury, such as periventricular leucomalacia show similar abnormalities.6
Interestingly, spectra obtained shortly after delivery from infants who have clearly suffered a severe episode of birth asphyxia are frequently normal, or almost so. Abnormalities develop over the first days of life, reaching a maximum at about 3 days, and at this stage intracellular pH (pHi) generally remains normal or may increase slightly. In surviving infants, recovery of the spectra to normal usually takes place over the succeeding 2 weeks or so, but there may then be a reduction in total phosphorus signal, indicating permanent loss of brain tissue.1
Recent studies of hypoxic-ischemic injury in the newborn piglet using high field strength magnetic resonance have demonstrated the sequence of events more clearly. When newborn piglets are subjected to a period of hypoxia-ischemia, there is a rapid fall in PCr/Pi, pHi and eventually ATP. If the animals are then promptly resuscitated, these alterations return to close to baseline values within an hour or two. After some 8 hours delayed alterations in PCr/Pi similar to those seen in infants suffering from post-asphyxial encephalopathy are seen, also occurring without a fall in pH.2
Proton Magnetic Resonance Spectroscopy
Recent work has begun to delineate the effects of birth asphyxia on1H magnetic resonance spectra. Using this technically demanding technique it is possible to observe changes in the concentrations of relevant species such as lactate, glutamate and glutamine, and n-acetyl aspartate (a compound almost exclusively found in neurons). Measurements in newborn infants have shown that lactate is present on the first day after birth in infants not suspected of cerebral injury, but that it is much higher in infants suffering birth asphyxia. High levels of lactate have been seen within 12 hours after birth asphyxia, and probably persist in infants with a poor neurological outcome.
The increase in cerebral lactate occurs before the delayed fall in PCr/Pi, and there is a linear relation between the magnitude of the rise in cerebral lactate and the severity of the later fall in PCr/Pi.10
Secondary Energy Failure
The mechanisms of this sequence of events are not clearly understood. The babies suffer an acute hypoxic- ischemic episode before delivery which produces the changes in the phosphorus spectra and pH1 observed in newborn animals. These changes may be designated primary energy failure. By the time the babies have been resuscitated and stabilized, and hence can be investigated, the31P spectra have reverted to normal. The episode of primary energy failure sets up a chain of reactions and vicious circles, that leads later to the secondary phase.9
This evolution may be regarded as indicative of delayed brain cell damage, a well known feature of neuropathology. The mechanisms involved are currently under intense study in numerous laboratories, because of the implications for stroke in adults, as well as for birth-asphyxiated babies. These considerations are dealt with in detail elsewhere in this symposium.
Prognosis
A series of 61 infants suspected of hypoxic-ischemic injury at birth were studied at one year of age. The extent to which PCr/Pi falls during secondary energy failure has been found to have prognostic implications. Infants progressing normally or with minor impairments of tone (of little significance) were found to have had normal values for PCr/Pi in the newborn period. Values from infants with isolated major neuromotor impairments were lower and those from infants with multiple major (and severe) impairments, lower still, not far above those from infants who died. Measurement of the Griffiths General Quotient in survivors showed a significant linear relation with the values for PCr/Pi.1 These results provide evidence for a continuum of reproductive casualty attributable to perinatal hypoxic-ischemic brain injury. It was also found that the positive predictive value for death or multiple major impairments for babies with values for PCr/Pi less than the 2.5th centile for normal babies was 93 percent, sufficiently high to be taken into account when deciding how far to pursue care in infants with similar findings.
Near Infrared Spectroscopy
Near infrared spectroscopy depends on the absorption of light in the near infrared region of the spectrum (700-1000 nm) as it traverses the head. Light at these wavelengths penetrates tissue far more readily than does visible light, so that with modern technology it is possible to make observations through heads with diameters up to 8 cm.
The technique was first clearly described by Jobsis in 1977.7 More recently methods have been worked out for quantifying a number of important cerebral variables. For example, changes in the global concentrations of the chromophores oxyhemoglobin, deoxyhemoglobin and oxidized cytochrome aa3 can be measured, cerebral blood flow and volume can be derived, and the reactivity of the cerebral circulation to changes in arterial carbon dioxide (PaCO2) can readily be tested.11-13 Continuous or repeated observations can be made in the neonatal unit without interference to normal nursing and medical procedures.
As stated earlier, infants who have suffered an acute reversed hypoxic-ischemic episode such as birth asphyxia frequently show normal phosphorus energetics shortly after delivery, but then go on to develop secondary energy failure. Recent data show that before secondary energy failure develops, cerebral abnormalities are often detectable by near infrared spectroscopy. For example, cerebral blood volume is raised and its carbon dioxide reactivity is much reduced.11 These abnormalities demonstrate vascular derangement during the development of secondary energy failure, but confirm other studies which have shown that cerebral substrate delivery in the period when secondary energy failure is developing is likely to be normal or increased.4
Electrophysiological Measurements
Many studies have shown that EEG with normal background activity in the first week after delivery is a reliable indicator of a good prognosis.8 One recent study of the changes in EEG in the first 4 hours after birth asphyxia showed that an abnormal recording had a high prognostic value for later neurodevelopmental impairment.
Studies in fetal lambs have also clearly shown that cerebral ischemia is followed by a reproducible pattern of abnormalities in the EEG power spectrum; a period of reduced intensity followed by a period of hyperactivity. The period of reduced activity correlated with histological outcome.5 Equally interesting are observations of alterations in cerebral impedance after cerebral ischemia. Cerebral impedance is increased if extracellular water volume decreases, and thus a failure of membrane pumps allowing cellular swelling is associated with increased cerebral impedance. In lambs subjected to a period of carotid artery occlusion in utero, cerebral electrical impedance follows a biphasic pattern. During the initial insult there is an increase in impedance which returns towards normal on resuscitation. A delayed increase follows some 8 to 12 hours later in the absence of further reductions in cerebral blood flow.5
The biphasic time course of electrophysiological events in fetal sheep closely parallel the findings of MRS, and reinforce the importance of secondary cellular injury occurring many hours after resuscitation from hypoxic-ischemic injury.
Conclusion
Non-invasive techniques have demonstrated that birth asphyxia involves at least two phases of cellular damage: a primary phase during substrate deprivation when CBF is probably low and the cells become acidotic, and a secondary phase which begins some 8 hours after resuscitation when CBF and pH1 are normal or high. Animal models used to investigate hypoxic-ischemic injury in the newborn period should only be accepted if evidence of a similar biphasic injury is available.
Acknowledgments
I am grateful for the invaluable help of Professor E.O.R. Reynolds, Dr. E.B. Cady and Dr. D. Wertheim.
References
- Azzopardi D, Wyatt JS, Cady EB et al. Prognosis of newborn infants with hypoxic-ischemic brain injury assessed by phosphorus magnetic resonance spectroscopy Pediatr Res 25:445-451, 1989.
- Lorek A, Takei Y, Cady EB et al. Delayed ("secondary") cerebral energy failure after acute hypoxia-ischemia in the newborn piglet: Continuous 48 hour studies by phosphorus magnetic resonance spectroscopy Pediatr Res 36:699-706, 1994.
- Edwards AD, Wyatt JS, Richardson C et al. Cotside measurement of cerebral blood flow in ill newborn infants by near infrared spectroscopy Lancet. 2:770-771, 1988.
- Frewen TC, Kissoon N, Kronick J et al. Cerebral blood flow, cross-brain oxygen extraction, and fontanelle pressure after hypoxic-ischemic injury in newborn infants. J Pediatr 118:265-271, 1991.
- Gunn AJ, Parer JT, Mallard EC et al. Cerebral histologic and electrocorticographic changes after asphyxia in fetal sheep. Ped Res 31:486- 491, 1992.
- Hamilton PA, Hope PL, Cady EB et al. Impaired energy metabolism in brains of newborn infants with increased cerebral echodensities. Lancet 1:1242-1246, 1986.
- Jobsis FF. Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science 198:1264- 1267, 1977.
- Takeuchi T, Watenabe K. The EEG evolution and neurological prognosis of perinatal hypoxia neonates. Brain Dev 11:115-120, 1989.
- Wyatt JS, Edwards AD, Azzopardi D et al. Magnetic resonance and near infrared spectroscopy for investigation of perinatal hypoxic-ischemic brain injury. Arch Dis Child 64:953-963, 1989.
- Hanrahan D, Azzopardi D, Manji K et al. Cerebral lactate concentration within 18 hours of birth asphyxia studied by1H magnetic resonance spectroscopy. Early Hum Dev (in press).
- Wyatt JS, Cope M, Delpy DT et al. Quantitation of cerebral blood volume in human infants by near-infrared spectroscopy. J AppI Physiol 68:1086-1091, 1990.
- Wyatt JS, Edwards AD, Delpy DT et al. Response of cerebral blood volume to changes in arterial carbon dioxide tension in preterm and term infants. Pediatr Res 29:553-557, 1991.
Clinical Research Methodology for Studies Based on Consensus Definition of Birth Asphyxia
John C. Sinclair, M.D.
McMaster University Medical Center, Hamilton,
Ontario, Canada
In reality, birth asphyxia is a research laboratory concept, not yet translatable into a clinical measure. All of our clinical measures are poor proxies for impaired gas exchange at the feto-placental level.1
Introduction
It is the focus of this meeting to re-examine Nigel Paneth's well justified nihilism and to attempt a definition of birth asphyxia. Mercifully, I have not been given the task of suggesting what the definition of birth asphyxia should be.2 Rather, the issue I have been asked to address is the following: assuming we had consensus on what constitutes a valid, reliable and clinically applicable definition of birth asphyxia, what questions would we ask, and what are the methodologic requirements for clinical research designed to answer those questions? Specifically, I will briefly consider four types of research questions:
- Evaluation of putative causes of birth asphyxia;
- Evaluation of diagnostic tests for birth asphyxia;
- Assessment of the natural history! prognosis of birth asphyxia; and
- Evaluation of treatment and prevention of birth asphyxia.
First, however, I would like to make two general points.
The Putative Causal Chain
I assume that the construct of birth asphyxia includes a critical reduction in fetomaternal gas exchange during labor and delivery which may lead to death, morbidity in the fetus/neonate and impaired neurodevelopmental outcome in infancy and childhood. However, I do not assume that birth asphyxia is either a necessary or sufficient cause of impaired outcome. In the NCPP,3 80 percent of infants weighing >2500g at birth and who developed cerebral palsy did not have 5-minute Apgar scores less than 7 (suggesting either that most cerebral palsy in term infants is not caused by birth asphyxia or that a low Apgar score is a poor measure of birth asphyxia). Moreover other factors may modify the tolerance to reduced fetomaternal gas exchange (suggesting that a specified reduction may not be sufficient in all cases to cause impairment or disability). Thus, we should design clinical research with the expectation that birth asphyxia is an important but perhaps modest contributor to the population burden of cerebral palsy and that it will be necessary to control for confounding arising from other determinants of adverse outcomes in infancy and childhood and from co-variates that may modify the relation between exposure and outcome.
Two Forms of "Gold Standard" Definition of Birth Asphyxia
What we require of a gold standard definition of birth asphyxia is that it be valid, able to be applied with good reproducibility, and clinically feasible. The types of research questions to be considered in this presentation suggest that two forms of gold standard definition of birth asphyxia will be needed: one for studies under highly controlled circumstances and one for field or population-based studies. The first definition will include measures of the construct which are state-of-the-art and which may be technologically sophisticated, invasive, and expensive. Such measures could be applied only in certain centers and only in highly-selected patients. The second definition will include measures of the construct which are routinely available, non-invasive and inexpensive. Such measures could be applied in large populations.
Studies of Etiology/Causation of Birth Asphyxia
With a gold standard definition of birth asphyxia in hand, studies of causation of birth asphyxia could be launched. Putative causal factors could be evaluated with respect to their role in causing birth asphyxia as specifically defined.
Valid clinical research for the determination of causation requires the following:4
- Evidence from true experiments or from cohort or case-control studies which control bias;
- Analysis of the strength of association between the putative causal factor and the outcome;
- Demonstration of the consistency of the association across studies;
- Demonstration that the temporal relationship is correct—i.e., that exposure precedes outcome; and
- Evaluation of a dose-response gradient.
The results are analyzed and reported as the relative risk (in randomized trials and cohort studies) or the odds ratio (in case-control studies, because the relative risk can not be calculated in such studies).
Putative causal factors which are identified and supported by such studies are classified as potentially remediable or not. Those that are potentially remediable can then be further studied in randomized trials of interventions designed to remove or ameliorate them (see section on Studies of Treatment/Prevention of Birth Asphyxia).
Studies of Diagnosis of Birth Asphyxia
With a gold standard definition of birth asphyxia in hand, diagnostic tests for birth asphyxia could be evaluated. Such tests include clinical data (such as the Apgar score), historical data (such as a history of meconium passage during labor) and laboratory examinations. In order to avoid circular reasoning, any components of the gold standard definition of birth asphyxia cannot themselves serve as diagnostic criteria.5 For example, hypoxic-ischemic encephalopathy could be considered as either part of a gold standard definition of birth asphyxia, or as a diagnostic criterion to be evaluated against a gold standard, but not both.
Valid clinical research for the evaluation of any diagnostic test requires the following:6
- An independent, blind comparison with the gold standard;
- A patient sample that includes an appropriate spectrum of patients to whom the diagnostic test will be applied in clinical practice (i.e., does the test distinguish between target disorders or states that might otherwise be confused?)
- Avoidance of work-up bias whereby the results of the test being evaluated influence the decision to perform the examinations which constitute the gold standard (this is a potential problem especially when the gold standard includes criteria ascertained using invasive or expensive tests); and
- Description of the methods for performing the test in sufficient detail to permit replication.
The diagnostic test results should be analyzed and reported using methods which display the properties of the test: sensitivity specificity, likelihood ratios associated with different levels of test result, and ROC curves which display the effect of shifting diagnostic thresholds. Conversely test results should not be analyzed and reported only for attributes which are highly prevalence-dependent and therefore are properties both of the test and of the patient sample in which the test is applied, i.e., predictive values, or using clinically inappropriate methods of analysis, e.g.,correlation and regression.
Diagnostic tests used in series pose a special problem if such tests are not independent (for example, it is usually unwarranted to assume independence of symptoms or signs within dlisease). With non-independence, it is not correct to assume that the post-test odds (or risk) following test one equals the pre-test odds (or risk) for test two. To do this leads systematically at the end of the diagnostic pathway to overestimation of the odds (or risk) of the target disorder. To avoid this, the diagnostic properties of the second test in sequence (i.e., sensitivity, specificity, likelihood ratios) should be calculated twice: in patients with a positive result and in patients with a negative result on the first test.7 Alternatively (and when more than two diagnostic tests are used in sequence) it is appropriate to analyze the results using logistic regression or the Spiegelhalter-Knill-Jones approach.8 In the latter method, the likelihood ratios from diagnostic test results are entered into a multiple logistic regression, and from this equation are derived adjusted weights of evidence which form a score which expresses the probability (based on all diagnostic test results) that the patient has the target disorder.
Studies of Prognosis of Birth Asphyxia
With a gold standard definition of birth asphyxia in hand, the prognosis of birth asphyxia could be studied. Outcomes of interest include both short-term outcomes (e.g., organ dysfunction, death in the neonatal period) and long-term outcomes (e.g., neurodevelopment, cerebral palsy, late death).
Valid clinical research for the study of prognosis requires the following:9
- A representative sample of patients at a uniform, well-defined point in the course of the disease/problem (an inception cohort). Obtaining a representative sample of birth asphyxia in the term infant poses a special difficulty. Most cases are born outside academic medical centers, and those born within such centers should not be assumed to be representative of the total population. Sampling strategies will need to be developed to obtain representative samples for prognostic studies. It is here that a simple gold standard definition of birth asphyxia, suitable for field studies, will have special application;
- Follow-up that is sufficiently long and complete. The required length of follow-up will be determined by the clinical outcomes of interest. For the diagnosis of major neurodevelopmental problems, follow-up to at least 18 months would seem to be the minimum requirement. For more subtle neurodevelopmental impairments, for intelligence testing, and for school learning problems, 5-7 years of follow-up will be required.
The problem of competing events has important application in the follow-up of birth asphyxia. Early death competes with later outcomes such as cerebral palsy in the sense that one cannot exhibit cerebral palsy if one has died soon after birth. To obtain an unbiased estimate of prognosis, therefore, the early and late outcomes should be separately reported by birth cohorts and, when appropriate, aggregated (e.g., either infant death or cerebral palsy).
The longer the follow-up which is required, the greater the risk of losing patients. Moreover, loss to follow-up should not be assumed to be random; rather, it may be selective and for reasons related to outcome. A study of prognosis is fundamentally compromised if the loss-to-follow- up rate is high in relation to the incidence of the adverse outcome(s) of interest. Thus, especially stringent procedures for maintenance of the inception cohort will be required in studies which seek to determine the incidence of late events which are relatively uncommon—e.g., cerebral palsy;
- The use of objective, unbiased outcome criteria: This will be particularly important for the measurement of outcomes that require judgment—e.g., cause of death, severity of impairment or disability grading of cerebral palsy. To avoid bias, the individual assessing such outcomes should be blind to the presence of absence of prognostic factors, e.g., severity of birth asphyxia; and
- Adjustment for important prognostic factors. In comparing the prognosis of two groups of patients, for example patients with and without birth asphyxia, it is important to adjust for other determinants of the outcome (e.g., socioeconomic class) that may be differently distributed in the two groups.
The prognostic study results should be analyzed and reported by providing the point estimate and confidence interval for each outcome of interest. In analyses which seek to associate the presence or absence of prognostic risk factors with the presence or absence of the target outcome and where there are multiple prognostic factors, the problem of non-independence of prognostic factors may arise (analogous to non- independence of diagnostic tests used in a series). To avoid overestimation of the probability of the target disorder arising from consideration of multiple prognostic factors which are non-independent, it is appropriate to analyze the results using logistic regression or the Spiegelhalter-Knill-Jones system,8 as discussed for studies of diagnostic tests.
Studies of Treatment/Prevention of Birth Asphyxia
With a gold standard definition of birth asphyxia in hand, the effectiveness of interventions for the treatment or prevention of birth asphyxia could be studied. In studies of treatment, the gold standard would be applied to select those babies who qualify for entry to the study; in studies of prevention, the gold standard would be applied to define the target outcome.
Valid clinical research for the study of treatment/prevention requires the following10:
- Random assignment of patient to treatment. There is at present a striking paucity of randomized trials of any aspect of treatment of babies who have presumed birth asphyxia.11 I believe this is due largely to uncertainty as to how to proceed ethically rather than to denial of the scientific need for randomized trials in this situation. Progress is needed in identifying ethically acceptable alternatives to prospective individual informed consent, such as (for example) seeking anticipatory authorization or seeking authorization after the fact,12 in order to facilitate the conduct of future randomized trials of treatment of birth asphyxia
- Complete follow-up, with patients analyzed in the groups to which they were randomized; and
- Blind assessment of outcome and, where possible, blinding of the intervention.
The results should be analyzed and reported using both relative and absolute estimators of treatment effect13—i.e., relative risk (and its complement, relative risk reduction) and absolute risk reduction. The inverse of the latter, i.e., 1/absolute risk reduction, defines the number needed to treat in order to expect to prevent one patient having the adverse target outcome. This is a particularly important measure of treatment effect when there are important costs of treatment—either clinical side effects or economic costs. For each of the treatment effect estimators described above the point estimate and a measure of precision (typically the 95 percent confidence interval) should be reported.
Can Clinical Research Validate A Definition of Birth Asphyxia?
A best case scenario resulting from the program of research outlined above would be:
- Causes of birth asphyxia (as defined) are identified, including some potentially preventable causes;
- Randomized trials of interventions to attack preventable causes are carried out which prove the effectiveness of the intervention in preventing birth asphyxia (as defined);
- Accurate diagnostic tests for birth asphyxia (as defined) are validated;
- Accurate description of the prognosis of birth asphyxia (as defined) is achieved, including both early and late outcomes;
- Randomized trials of treatment of birth asphyxia (as defined) show effectiveness in preventing early and late adverse outcomes; and
- At the population level, this research evidence is efficiently translated into practice (with no stepdown of effectiveness because of lack of coverage or noncompliance) and the population burden of birth asphyxia (as defined) is reduced.
To the extent that such a scenario unfolds, the criteria of the scientific method are met. In this global sense, criterion validity is established.
References
- Paneth N. The causes of cerebral palsy. Recent evidence. Clin Invest Med 16:95-102, 1993.
- Hull J, Dodd K. What is birth asphyxia? Br J Obstet Gynecol 98:953-955, 1991.
- Nelson KB, Ellenberg JH. Apgar scores as predictors of chronic neurologic disability. Pediatrics 68:36-44, 1981.
- Department of Clinical Epidemiology and Biostatistics, McMaster University Health Sciences Centre. How to read clinical journals: IV. To determine etiology or causation. Can Med Assoc J 124:985-990, 1981.
- Wasson JH, Sox HC, Neff RK, Goldman L. Clinical prediction rules. Application and methodologic standards. N Engl J Med 313:793-799, 1985.
- Jaeschke R, Guyatt G, Sackett DL. Evidence-Based Medicine Working Group. Users' guides to the medical literature. III. How to use an article about a diagnostic test. A. Are the results of the study valid? JAMA 271:389-391, 1994. B. What are the results and will they help me in caring for my patients? JAMA 271:703-707, 1994.
- Sox HC Jr. Probability theory in the use of diagnostic tests. An introduction to critical study of the literature. Ann Int Med 104:60-66, 1986.
- Seymour DG, Green M, Vaz FG. Making better decisions: Construction of clinical scoring systems by the Spiegelhalter-Knill-Jones approach. Br Med J 300:223-226, 1990.
- Department of Clinical Epidemiology and Biostatistics, McMaster University Health Sciences Centre. How to read clinical journals. III. To learn about the clinical course and prognosis of disease. Can Med Assoc J 124:869-872, 1981.
- Guyatt GH, Sackett DL, Cook DJ. Evidence-Based Working Group. Users' guides to the medical literature. II. How to use an article about therapy or prevention. A. Are the results of the study valid? JAMA 270:2598-2601, 1993. B. What were the results and will they help me in caring for my patients? JAMA 271:59-63, 1994.
- Sinclair JC, Bracken MB (eds). Effective Care of the Newborn Infant. Oxford: University Press, 1992.
- Consent Panel Task Force of the National Council on Bioethics in Human Research, Canada. Report on Research Involving Children, Special Circumstances, pp. 45-49, 1991.
- Bracken MB, Sinclair JC. Clinically useful measures of effect in binary analyses of randomized clinical trials. J Clin Epidemiol 87:881- 889, 1994.
Discussion
William Oh, M.D.
Women and Infants Hospital of Rhode Island, Providence
There is a real need for future research in the area of perinatal asphyxia with reference to diagnosis, prognosis and intervention. I would agree with Jack Sinclair that a good starting point is to arrive at a consensus as to what constitutes perinatal asphyxia. My own bias is that we may need a good animal model that will allow us to establish the pathophysiology of perinatal asphyxia. The animal model will allow for direct assessment and establish correlation between injury producing placental fetal compromise and histologic and physiologic interpretation of primary outcome variables in the fetus and neonates. The next step is to utilize highly sophisticated non-invasive technologies such as those described by Professor Edward that will allow us to further explore the clinico-pathological correlation based in part on the findings obtained in the animal model.
Research endeavors to provide data for prognosis and to test the effects of interventions are sorely needed. Clinical research is probably the most appropriate approach in obtaining the data. In addition to arriving at a consensus on the definition of perinatal or birth asphyxia, there is a need to define a priori the varying degrees of severity of perinatal asphyxia in assessing the short and long term outcomes. In developing a research protocol to assess the long term outcome variables (quality of life of the survivors), it is essential that multitudes of potential confounding variables be taken into account. These varlables include: socioeconomic status, ethnicity language barriers, intercurrent illness, behavioral or psychosocial interventions, just to name a few. Of course, it is absolutely essential that the follow-up duration and compliance rate be kept optimal to lend credibility to the data.
Discussion
Jerold F. Lucey, M.D., F.A.A.P.
Department of Pediatrics, Medical Center Hospital of Vermont, Burlington
Introduction
Professor Edwards has summarized and reviewed the current status of three of the newer non-invasive methodologies for documenting and timing the evidence of brain asphyxia in the prenatal period. The human clinical experience with these devices is, to date, small. The good news is that it is quite promising. It would appear that abnormalities observed develop over the first few days of life. This suggests the possibility that some therapeutic intervention might be possible and that its effects could be followed by serial studies. The concept of an insult producing a primary energy failure which leads to a secondary phase of delayed cell damage is an exciting one. It means we may have a chance to do something. Neuroprotective drugs may actually have a future.
Early studies also indicate that it may be possible to prognosticate the degree of brain damage in survivors. If that holds up, it means that proper randomized controlled studies can be carried out. They won't be easy, but they can and should be done.
Dr. Sinclair's presentation indicates how these clinical trials should be done. This field has become almost as complex and demanding as the newer non-invasive neurophysiological study techniques. Large numbers of patients are required to prove the safety and effectiveness of new therapies.
Can these two fields of research ever be combined to meet at the birth of an asphyxiated infants? The discouraging answer is that this just won't happen unless we have some major changes in the way research is carried out in these fields.
Changes Needed
The first thing our neurologic and imaging colleagues need to do is develop a practical device which would allow clinicians to make an accurate early diagnosis of perinatal brain damage. I would give them 10 years to do this. Figure 1 outlines the specifications for such a dream machine.
It should be small, easy to use and simple to interpret. It must be rugged, inexpensive and give rapid results. It must be able to be used repeatedly prenatally and during labor.
Once this dream machine is available, we will have a few other problems. We will need a set of definitions (the gold standard). Then we'll need to organize a series of proper, randomized controlled trials. I suggest that this effort be led by a group of professional trialists. I think this is very important. As an editor I've seen too many reviews of large trials designed by well-intentioned investigators, rejected because the trial was seriously flawed. I would give this group 5 years to design proper trials.
It seems to me worthwhile to start on the design of such trials now. Several promising neuroprotective drug trials are now well advanced in adults. The NICHD should start work now with the FDA and private industry on the design of future trials in infants sharing knowledge, expertise and expenses.
FIGURE 1: The "Dream Machine" for Diagnosis of Perinatal Brain Damage
Small
Simple to Use
Inexpensive
Rugged
Non-Invasive
Rapid Results
Non-ionizing radiation
Tell time and etiology of injury
Repeated studies
The next problem is a source of patients. Unfortunately the majority of seriously brain damaged infants are not born in university medical centers. They are often born in smaller hospitals and hours later transported to university research centers. This is a major problem. If the dream machine was available, it could be used in smaller hospitals. If these smaller hospitals were joined together in a project several sequential, large pragmatic trials could be carried out. We would arrive at an answer much sooner than if we wait for 10-30 years for each university group to design, fund and carry out its own small trial.
What I am proposing, as the way to go, is a Manhattan Project/Space Program approach rather than the present non-system we are trapped in. To me, the goal, which is the elimination of perinatal brain damage, is just as important as these previous other successful expensive government projects.
It will need intellectual and political leadership. Somebody (NICHD) needs to take the leadership role, cut across specialty boundaries and get the project started. We're probably the only country in the world capable of doing this. I think it's within our country's capacities. The time to start planning is now.
 BACK TO TOP
BACK TO TOP