
NIH-funded study finds that gene appears to regulate protein signals inside the cell
Researchers working with mice have uncovered a potential clue to polydactyly—a birth defect involving extra fingers on the hand or extra toes on the feet. The researchers have found that a mouse version of polydactyly results from a malfunction of the cellular machinery that processes one of the cell's internal transportation vehicles. The Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), part of the National Institutes of Health, provided funding for the study.
Polydactyly is estimated to occur in roughly 1 of every 500 to 1,000 births. The condition appears to result from a mutation in any of a number of genes. Polydactyly may involve only one extra finger or toe, or it may include multiple extra digits. The condition may also appear as part of a larger disorder having other organ abnormalities, some of which may be serious or life- threatening.
The study, published online in Cell Reports, was undertaken by Licia Selleri, M.D., Ph.D., Weill Cornell Medical College, New York City, and colleagues at Cornell and other institutions.
"Dr. Selleri and her colleagues have discovered that an unexpected cellular pathway is involved in the generation of extra digits," said Lorette Javois, Ph.D., of the Developmental Biology and Structural Variation Branch at NICHD. "The findings expand what we know about genetic pathways involved in normal limb development and offer researchers a new direction in which to search for human mutations."
Additional funding was provided by NIH's National Institute of Dental and Craniofacial Research.
The researchers found that a mouse form of polydactyly appears to result from an error in a single gene out of a group containing the information needed to make a protein complex. The protein complex is called Endosomal Sorting Complex Required for Transport II—ESCRT-II, for short. Endosomes ferry molecules from the cell's surface to the cell's interior. When it functions normally, ESCRT-II processes endosomes for eventual disassembly within the cell. The mutation impairs the functioning of ESCRT-II, endosomes cannot be processed properly. As a result, an excess of a hormone known as fibroblast growth factor accumulates on the cell's surface.
"Since this congenital defect appears both in isolation and in conjunction with other abnormalities, gaining fundamental knowledge of these genetic pathways is vital to developing effective genetic diagnostic screens and directed therapies," Dr. Selleri said.
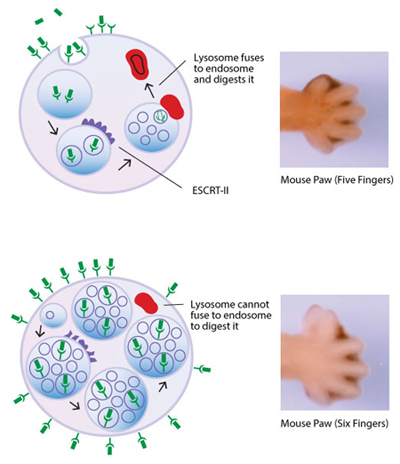
The fibroblast growth factors are a family of proteins known as cell signaling molecules.
At top left: After a fibroblast growth factor binds to a cell, it triggers two potential sequences of events. Under typical conditions, it sets off a cascade of chemical reactions that eventually cause cells to divide, and even to differentiate into tissues and organs. In the other, shown in the image at the above left, it's processed and then marked for eventual disassembly. With this internal breakdown process, the cell achieves a balance—once the cell acquires a critical mass of a fibroblast growth factor, the excess is marked for elimination. The process begins with the fibroblast growth factor (FGF) attaching to a special site, or receptor, on the cell surface, something like the way a key fits into a lock. The receptor changes shape, setting off the reactions that trigger the cell into such actions as dividing or giving rise to an organ or tissue type. At some point, sufficient FGF has bound to receptors on the cell surface. In response, pocket-like structures form on the cell's surface. The cell surface changes shape—it involutes, like the surface of a water-filled balloon with a finger in it. This sunken-in structure contains many such FGF-receptor pairs. Eventually, this involution pinches off inside the cell, forming a spherical structure known as an endosome. Like a bathysphere ferrying ocean researchers to the sea depths, the endosome conveys the FGF-receptor duplexes deep into the cell. During their journey, the endosomes encounter a protein complex, called Endosomal Sorting Complex Required for Transport II—ESCRT-II, for short. When it's functioning normally, ESCRT-II processes the endosome, reducing it in size. The details of just how ESCRIT-II processes the endosome aren't fully understood. Once this processing takes place, the endosome fuses with another cellular body, called a lysosome. The lysosome disassembles the endosome, degrading it and its contents—FGF and its receptor.
At bottom left: The researchers discovered that, in the mouse strain they bred for the study, a mutation in a gene for a key subunit of ESCRT-II impairs the ability of ESCRT-II to do its job. This gene contains the information needed to make vacuolar protein sorting protein 25, or Vps25. The mutant Vps25 protein interferes with ESCRT-II's ability to do its job. Because the endosomes aren't sufficiently processed, the lysosomes have trouble breaking them apart. As a result, endosomes accumulate inside the cell. Fewer endosomes form at the cell surface, and FGF-receptor pairs accumulate on the surface because they can't be removed rapidly enough. This situation creates an imbalance—increasing the amount of FGF bound to receptors on the cell surface. The end result in the mice with the Vps25 mutation is polydactyly.
###
About the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD): The NICHD sponsors research on development, before and after birth; maternal, child, and family health; reproductive biology and population issues; and medical rehabilitation. For more information, visit the Institute's website at http://www.nichd.nih.gov/.
About the National Institutes of Health (NIH): NIH, the nation's medical research agency, includes 27 Institutes and Centers and is a component of the U.S. Department of Health and Human Services. NIH is the primary federal agency conducting and supporting basic, clinical, and translational medical research, and is investigating the causes, treatments, and cures for both common and rare diseases. For more information about NIH and its programs, visit http://www.nih.gov.
 BACK TO TOP
BACK TO TOP