
Defects in lysosome acidity can lead to neurodegenerative disease
The misdelivery of proteins to lysosomes, the structures within cells that break down unneeded or worn-out proteins, fats, carbohydrates and cell parts, as well as viruses and bacteria, appears to underlie a family of rare neurodegenerative diseases, according to a study led by researchers at the National Institutes of Health. A pump called v-ATPase, which regulates the acidity of lysosomes, requires a special tagging and delivery process called palmitoylation. When this process is disrupted, lysosomes are no longer acidic, which can lead to neurodegenerative diseases called lysosomal storage disorders. Finding ways to restore this tagging process may result in new therapies for these disorders. The study appears in the March 7, 2017, issue of Nature Communications.
Background
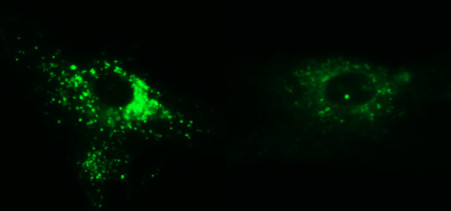
Lysosomes break down unneeded elements within a cell. They contain digestive enzymes that are active only in an acidic environment. Therefore, lysosomes must become acidic in order to function. A pump called v-ATPase, which is made up of many proteins and spans the lysosomal membrane, brings in molecules that create an acidic environment.
Researchers know that when lysosomes cannot become acidic, lysosomal storage disorders can occur. These disorders are caused by a toxic buildup of undigested proteins and cellular components in neurons. Once such disorder called infantile neuronal ceroid lipofuscinosis (INCL) begins in infancy and causes intellectual disability, seizures, and problems walking, speaking, and seeing. There is no cure, and most patients do not survive past childhood.
Prior to the new study, researchers knew that INCL was caused by genetic mutations that impair the function of a lysosomal enzyme called PPT1. However, scientists did not understand exactly how a deficiency in PPT1 leads to disease development.
Results
Researchers led by Anil Mukherjee, M.D., Ph.D., of NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development, studied a mouse model of INCL, which lacks the PPT1 enzyme. They discovered that V0a1, which is part of the v-ATPase pump, was virtually undetectable on the lysosomal membrane—its normal location. The team found that in order for v-ATPase to be delivered to lysosomes, V0a1 needed to undergo a special tagging process called palmitoylation. This process relied on PPT1. In the case of INCL mice, V0a1 was misrouted to the neuron’s surface.
Next, the researchers treated INCL mice with the drug NtBuHA, which mimics PPT1 activity. The drug restored V0a1’s presence in lysosomes when compared to untreated mice. Furthermore, lysosomes from the neurons of treated mice also had near-normal acidity.
Significance
The study shows that palmitoylation is required for delivering v-ATPase to lysosomes and helps explain the lysosome’s low acidity in INCL.
“Impaired lysosome function is implicated in more common neurodegenerative disorders, such as Alzheimer’s and Parkinson’s, although the precise mechanism of the defect remains unclear,” said Dr. Mukherjee. The lead author of the article, Maria Bagh, Ph.D., added, “Small molecules mimicking PPT1 activity may be an effective therapeutic target to restore function in lysosomal storage disorders, as well as in other neurodegenerative diseases.”
More research is needed to explore the feasibility and safety of targeting PPT1 with drugs and to clarify the role of PPT1 in other neurological diseases.
Reference
Bagh MB, Peng S, Chandra G, Zhang Z, Singh SP, Pattabiraman N, Liu A, and Mukherjee AB. Misrouting of v-ATPase subunit V0a1 dysregulates lysosomal acidification in a neurodegenerative lysosomal storage disease model. Nature Communications DOI: 10.1038/ncomms14612 (2017)
 BACK TO TOP
BACK TO TOP