
Findings in mice suggest new ways to treat long-term pain without opioids
Credit: NIH
Long-term pain and inflammation that occurs after nerve injury is triggered by a specific protein, according to a study in mice led by NIH’s Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) and the National Center for Complementary and Integrative Health (NCCIH). Identifying the role of the protein, dual leucine zipper kinase (DLK), may inform new ways to manage pain that that do not require opioids. The study appears in the July 3, 2018, issue of eLIFE.
Background
Opioids are often prescribed to treat pain following injury or surgery. However, misuse of opioids can lead to addiction and even death by drug overdose. NIH research seeks to identify new, non-addictive ways to manage pain.
The ability to feel pain comes from underlying mechanisms within the nervous system. However, when pain persists, it can become a debilitating health condition. Scientists do not fully understand how this type of chronic pain develops.
Results
The study was led by Claire Le Pichon, Ph.D., who heads NICHD’s Unit on the Development of Neurodegeneration, and Alexander Chesler, Ph.D., who leads NCCIH’s Section on Sensory Cells and Circuits. To explore the potential role of DLK in the development of pain, the team developed mice that lack DLK and compared how these animals and normal mice recovered from a nerve injury. They found that mice missing DLK did not develop long-term pain after injury, nor did their spinal cords develop a hallmark of nerve inflammation called microgliosis.
Next, the study team neutralized the effects of DLK by targeting it with a drug (GNE-3511). Mice that could produce DLK were treated with the drug twice daily after nerve injury. The animals showed limited evidence of long-term pain or subsequent microgliosis (videos of microscopy images of spinal cords from GNE-3511 treated and untreated mice are available at the eLIFE website).
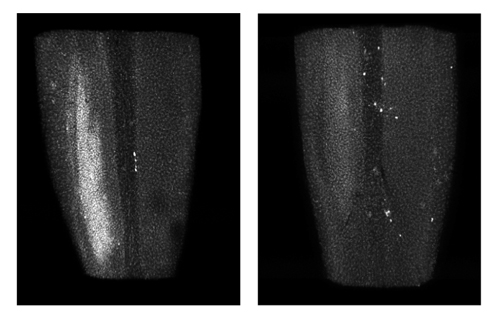
Damage to the spinal cord can develop hours to days after nerve injury. Normal mice (left) develop this damage (white patch), called microgliosis, while mice missing DLK (right) do not develop microgliosis.
Credit: NIH
The researchers discovered that in neurons, DLK regulates a set of genes associated with the development of pain, including the gene Csf1. Scientists knew that Csf1 is necessary for injury-related pain and microgliosis in mice, but they did not how this gene was regulated. The researchers propose that DLK functions as a so-called master regulator, coordinating a genetic program that results in pain and other injury-related damage.
Significance
The work provides new insight into how long-term pain develops after nerve injury.
“We hope that understanding these pathways will lead to new therapeutic approaches and more effective treatments for pain.” said Dr. Chesler.
“Additionally, our findings extend beyond nerve injury to other disorders, such as neurodegenerative diseases, where neuroinflammation also occurs,” added Dr. Le Pichon. “DLK may be similarly responsible for a neuronal signal to inflammatory cells in these contexts, and more research is needed to explore this possibility.”
Reference
Wlaschin JJ, Gluski JM, Nguyen E, Silberberg H, Thompson JH, Chesler AT, and Le Pichon CE. Dual leucine zipper kinase is required for mechanical allodynia and microgliosis after nerve injury. eLIFE DOI: 10.7554/eLife.33910.001 (2018)
 BACK TO TOP
BACK TO TOP