What is the incidence and prevalence of PKU and other forms of hyperphenylalaninemias, and what is known about the genetic and clinical variability?
Incidence and Prevalence of PKU and Hyperphenylalaninemia
Newborn screening for PKU began in 1961 and initial results were reported two years later by Guthrie and Susi (1963). In the first 2 years, 400,000 infants were tested in 29 states and 39 cases of PKU were found--an incidence of about 1 per 10,000. More recently, the American Academy of Pediatrics Policy Statement regarding Newborn Screening cites the incidence of PKU as 1 in 10,000 to 1 in 25,000 in the United States based on data from 1992 published by the Council of Regional Networks for Genetics Services (PDF - 192 KB) (American Academy of Pediatrics, 1996).
In an attempt to update the incidence of PKU in the United States, data from the Council of Regional Networks for Genetics Services, National Newborn Screening Report – 1994 (Newborn Screening Committee, 1999) was reviewed. CORN collected and summarized newborn screening data from each state and province (U.S. Virgin Islands, Puerto Rico, and Washington D.C.). During 1994, the number of live births in the United States was estimated at 4,020,306. Each racial group identified contributed to the newborn population as follows: White-66.5%; Hispanic-16%; Black-13.3%; Asian or Pacific Islanders-3.3%; Native Americans-0.8%. Of these live births, the report estimates that 3,807,187 newborns underwent an initial newborn screening test.
The CORN Report (Ibid.) provided extensive data regarding newborn screening, but there are problems with inconsistencies in data collection and reporting. For example, the number of infants born in Florida and Georgia is given, but the number of newborns screened is not given. Without these data, it is not possible to estimate the incidence of PKU in these two states. Other inconsistencies in reporting of data include failure to report sex and/or race of some newborns with PKU; failure to collect data regarding cases of hyperphenylalaninemia by some states; and failure to separate data gathered from first screening tests and second follow-up tests. The net result is an inaccurate tally of the newborns detected with PKU.
A potentially confounding problem in estimating the incidence of PKU is the wide variation in diagnostic criteria from one state to another. The definitions used for identification of classical PKU and clinically significant variant hyperphenylalaninemia are based on blood phenylalanine concentration and other diagnostic criteria (Tables 1 and 2). Most states (39%) report classical PKU as defined by blood phenylalanine > 20 mg/dL. Four states report blood phenylalanine values > 4 mg/dL and 3 states report values > 15 mg/dL. Another 26% of states rely on a diagnosis by specialist (no specific biochemical parameters defined in the report). In reporting clinically significant variant hyperphenylalaninemia, 30% of states rely on diagnosis by a specialist, whereas the blood phenylalanine criteria used in the remaining states range from ³ 4 mg/dL through £ 20 mg/dL. It logically follows that the variation in definitions used by the states to report classical PKU versus hyperphenylalaninemia variants could, of itself, alter estimates of the incidence of PKU cases from state to state.
Table 1. Screening Definitions for Classical Phenylketonuria
| Definitions for Classical PKU | No. of States or Provinces Using Definition (53 Total) |
|---|---|
| Blood phenylalanine >20 mg/dL | 22 |
| Dx by specialist, no specific parameters given | 14 |
| Blood phenylalanine >10 mg/dL | 4 |
| Blood phenylalanine > 4 mg/dL | 3 |
| Blood phenylalanine > 15 mg/dL | 2 (VT, OK) |
| Phenylalanine hydroxylase deficiency | 1 (KS) |
| Blood phenylalanine >16.5 mg/dL | 1 (NE) |
| Blood phenylalanine >8 mg/dL | 1 (KY) |
| Blood phenylalanine >6 mg/dL | 1 (NV) |
| Blood phenylalanine >2 mg/dL | 1 (LA) |
| Hyper-Phenylalanine definition | 1 (OR) |
| Not reported, Not available | 2 (VI, AK) |
Table 2. Screening Definitions for Clinically Significant Variant Hyperphenylalaninemia
| Definitions | No. of States and Provinces Using Definition (53 Total) |
|---|---|
| Diagnosis by Specialist (no criteria reported) | 16 |
| Blood phenylalanine >=10 to < 20 mg/dL | 8 |
| Blood phenylalanine > 6 mg/dL | 5 |
| Blood phenylalanine >=4 to < 10 mg/dL | 3 |
| Blood phenylalanine 10 to 12 mg/dL | 2 |
| Not reported, not available | 3 |
| Other Variations | 14 |
Overall blood phenylalanine range: = 4mg/dL to = 20 mg/dL
After 3,807,187 initial screening tests, the number of newborns reported as confirmed with classical PKU was 217, approximately 0.0057% of the newborns screened or 1:17,544. Annotation of the data indicates that this total does not include all of the states. This number included cases from Florida and Georgia although the number of individuals screened in these states were not reported (Virgin Islands not included). There were 16 cases of classical PKU reported for Florida and Georgia. Using the actual number of PKU cases, 217, and excluding cases reported for Florida and Georgia (16 cases) reduces the number of cases to 201. This changes the incidence of classical PKU to 0.0053% of newborns initially screened or 1:18,941. After initial screens, in the 26 states reporting cases, the number of newborns confirmed with clinically significant hyperphenylalaninemia variants was 79. This number represents approximately 0.0021% of the newborns screened or 1:48,192. Eight states were not included or did not collect these data
The method used for incorporating data for second screening tests is not clear. The total number of newborns with classical PKU after first and second screening tests adds up to 191 (Table 3); however, it was reported as 234 in the 1994 CORN Report. The discrepancy was explained as a problem with reliability in reporting of data. There are problems regarding the mode used by states for reporting multiplicity of data because first and second screening tests were not separated, and in some instances, the race and sex of the infant with PKU were not recorded.
Table 3. Incidence of Classical PKU Detected by Neonatal Screening According to Race
* The number of newborns screened in each racial group was estimated using the percentage of the total number of newborns belonging to each racial group and the number of newborns actually screened.
**The incidence was calculated for each racial group based on the number of initial screening tests even though the number of cases by race was reported after first and second screens.
aSome states counted Hispanics in other racial categories
| Race | PKU Infants Detected | Estimated Number of Newborns Screened* | Incidence** |
|---|---|---|---|
| White | 150 | 2,531,779 | 1:16,878 |
| Black | 4 | 506,355 | 1:126,588 |
| Hispanic a | 10 | 609,149 | 1:60,914 |
| Asian, Pacific Is. | 0 | 125,637 | |
| Native American | 2 | 30,457 | 1:15,228 |
| Other | 6 | ? | |
| Unknown | 24 | ? | |
| Total | 191 | 3,807,187 | 1:19,932 |
Using the number of initial screening tests and the racial distribution of infants born in the U.S., the cases of classical PKU distributed by race are presented in Table 3. States not reporting race were SD, NH, NV, MS, FL, and NE, accounting for 24 individuals with PKU. The majority (78.5%) of infants with PKU were white, and the estimated incidence for this racial group was considerably higher than for African Americans and Hispanics. Because the data used to generate Table 3 represent first and second screening tests, there is a slight discrepancy in the overall incidence for PKU (0.00501% or 1:19,932) compared to the previous estimate (0.0053% or 1:18,941). Both overall incidence estimates, however, are within the wide range cited by the American Academy of Pediatrics Policy Statement (American Academy of Pediatrics, 1996) regarding Newborn Screening that reports the incidence of PKU as 1 in 10,000 to 1 in 25,000 in the United States.
Amongst the sexes, the incidence of PKU is equally divided ( Table 4). In 1994, there were 98 males and 93 females, but the sex of 26 other individuals was not reported.
Table 4. Cases of Classical PKU Detected by Sex
| Sex | No. Detected | Percent of Total Individuals Detected |
|---|---|---|
| Male | 98 | 45.1 |
| Female | 93 | 42.8 |
| Not Reported | 26 | 11.9 |
| Total | 217 |
In an effort to answer the question 'Does variation in the definition used to identify individuals with classical PKU affect the reported incidence of PKU?', incidence data were summarized according to the diagnostic criteria used by each state. Table 5 cites the states detecting cases of PKU in 1994 and includes the definitions used for identifying those cases. Only states reporting cases of classical PKU and the number of individuals screened initially were used to generate incidence data. A total of 210 individuals with classical PKU were detected when reported by state on first and second screens.
Of the 42 states reporting individuals identified with classical PKU, 15 used the definition of >20mg/dL. Ten states used definitions in the range >2 mg/dL to >16.5 mg/dL. Fifteen states gave no specific parameters or relied on diagnosis by a specialist. Overall, there also does not appear to be any correlation between the specific biochemical parameter used by states and the number of PKU cases detected or the incidence of PKU, that is to say, states using more stringent parameters with a lower phenylalanine cutoff did not necessarily detect more cases
Eighty-five cases of clinically significant hyperphenylalaninemia variants were detected by the states on first and second screening tests. Table 6 cites the states detecting the hyperphenylalaninemia cases in 1994 and includes the definitions used for identifying them. Only states reporting cases and the number of individuals initially screened were used to generate incidence data. Of these 25 states, 10 used a definition of blood phenylalanine between 10 and 20 mg/dL or had an upper limit of 20 mg/dL. Eight states gave no specific diagnostic parameters or relied on a diagnosis by a specialist. There does not appear to be any correlation between the specific biochemical criteria used by states for screening and the number of cases detected.
Table 5. Cases of Classical PKU Detected by States on Initial and Second Screens Grouped According to Phenylalanine Diagnostic Definition
a Race and sex not given for 7 of the 15 cases reported. bRace and sex not given for 1 of the 4 cases reported. cRace and sex not specified. dRace and sex not given for 3 of the 13 cases reported. NSP: No specific parameters given. PAH-: Phenylalanine hydroxylase inactivity. CSH: Defined as Clinically significant Hyperphenylalaninemia. States/territories not detecting cases of Classical PKU: AL, CO, DE, DC, FL, HI, ND, VT, WY, VI. Data from Georgia are not included because the number of individuals screened was not reported.
| PKU Diagnostic Definition | State | PKU Cases Detected No./ Incidence | PKU Diagnostic Definition | State | PKU Cases Detected No./ Incidence |
|---|---|---|---|---|---|
| >= 20 mg/dL | AR | 1 / 1:33,280 | > 6mg/dL | NV | 4 c / 1:6,149 |
| >= 20 mg/dL | IA | 2 / 1:18,689 | > 4 mg/dL; Dx | ID | 1 / 1:19,314 |
| > 20 mg/dL | CT | 3 / 1:15,880 | >= 4 mg/dL | IN | 7 / 1:11,745 |
| > 20 mg/dL | ME | 3 / 1:4,733 | >2 mg/dL | LA | 2 / 1:33,901 |
| > 20 mg/dL | MD | 4 / 1:17,104 | Dx, NSP | CA | 16 /1:35,333 |
| > 20 mg/dL | MI | 15 a/ 1:9,093 | Dx, NSP | MA | 9 / 1:9,432 |
| > 20 mg/dL | MN | 2 / 1:32,994 | Dx, NSP | MO | 4 b/ 1:19,217 |
| > 20 mg/dL | MS | 2 / 1:20,695 | Dx, NSP | IL | 9 / 1:21,020 |
| > 20 mg/dL | MT | 1 / 1:10,858 | Dx, NSP | NH | 1 c / 1:14,603 |
| > 20 mg/dL | TN | 1 / 1:80,458 | Dx, NSP | NJ | 6 / 1:18,668 |
| > 20 mg/dL | TX | 10 / 1:36,758 | Dx, NSP | NY | 16 / 1:17,375 |
| > 20 mg/dL | UT | 3 /1:12,910 | Dx, NSP | NC | 7 / 1:14,283 |
| > 20 mg/dL | WV | 2 / 1:11,503 | Dx, NSP | OH | 13 d / 1:12,858 |
| > 20 mg/dL | WI | 8 / 1:8,400 | CSH | OR | 6 / 1:7,401 |
| > 20 mg/dL | PR | 2 / 1:30,704 | Dx, NSP | PA | 10 / 1:15,565 |
| > 16.5 mg/dL | NE | 3 c/ 1:7,712 | Dx, NSP | RI | 2 / 1:7,000 |
| >= 15 mg/dL | OK | 3 / 1:16,800 | Dx, NSP | SC | 5 / 1:8,538 |
| >= 10 mg/dL | AZ | 4 / 1:21,581 | Dx, NSP | SD | 1 c / 1:10,577 |
| > 10mg/dL | WA | 5 / 1:14,281 | PAH- | KS | 3 / 1:15,200 |
| > 10 mg/dL | NM | 3 / 1:9,394 | Not Available | AK | 2 / 1:5,392 |
| >8 mg/dL | KY | 5 / 1:9,981 | Phe elevated; NSP | VA | 4 / 1:24,772 |
Table 6. Cases of Clinically Significant Hyperphenylalaninemia Detected by States on Initial and Second Screens Grouped According to Phenylalanine Diagnostic Definition
a Race and sex not reported. States/territories not reporting cases of Clinically Significant Hyperphenylalaninemia Variants: AL, AK, AZ, DE, DC, FL, HI, IA, KS, LA, ME, MA, MT, NE, NV, NJ, NM, RI, SC, SD, UT, VT, VA, WV, PR, VI
| Diagnostic Definition | State | Cases Detected No./ Incidence |
Diagnostic Definition | State | Cases Detected No./ Incidence |
|---|---|---|---|---|---|
| 10-20 mg/dL | CT | 1 / 1:47,642 | > 6 mg/dL | ID | 2 / 1:9,657 |
| 10-20 mg/dL | MI | 9 / 1:15,155 | 6-10 mg/dL | WA | 1 / 1:71,406 |
| 10-20 mg/dL | MS | 2 / 1:20,695 | 4-10 mg/dL | WI | 1 / 1:67,200 |
| 10-20 mg/dL | TN | 2 / 1:40,229 | Dx; NSP | IL | 2 / 1:94,591 |
| 10-19.9 mg/dL | TX | 4 / 1:91,895 | Dx; NSP | IN | 6 / 1:13,702 |
| 20 mg/dL | KY | 3 / 1:16,636 | Dx; NSP | MO | 1 / 1:76,868 |
| >2 mg/dL | CO | 2 a / 1:29,132 | Dx; NSP | NH | 1 a / 1:14,603 |
| 8-20 mg/dL | MD | 2 / 1:34,208 | Dx; NSP | NC | 4 / 1:24,995 |
| 10-12 mg/dL | WY | 1 / 1:5,853 | Dx; NSP | ND | 1 / 1:9,820 |
| 6-15 mg/dL | OK | 1 / 1:50,401 | Dx; NSP | OH | 3 a / 1:55,720 |
| 4-20 mg/dL | MN | 2 / 1:32,994 | Dx; NSP | PA | 5 / 1:31,131 |
| 4 -10 mg/dL | AR | 1 / 1:33,280 | Not available | CA | 23 / 1:24,579 |
| >6 mg/dL | OR | 2 / 1:22,203 |
Other Estimates of the Incidence of Classical PKU
A review by Thalhammer (1975) cites extensive data for several European cities and/or countries encompassing the time period from 1960 to 1975. The frequencies of PKU and hyperphenylalaninemia differ from country to country and even within regions of the same country. Data cited for Boston in 1974 estimates the incidence of PKU at 1:13,914. The estimate for Portland is based on data from individuals screened between 1962 and 1974/6 and is given as 1:11,620. The incidences of hyperphenylalaninemia for Boston and Portland during the same time periods are 1:17,006 and 1:33,700 respectively. On the basis of 10 years of Maryland newborn-screening data, Hofman et al. (1991) concluded that the incidence of PKU in U.S. African Americans is about 1 in 50,000, or one-third that in whites.
PKU is rare in Ashkenazi Jews (Cohen et. al., 1961; Centerwall and Neff, 1961). Carter and Woolf (1961) noted that of the cases seen at that time, a large number had predecessors born in Ireland or West Scotland. The incidence at birth in northern Europeans may be about 1 per 10,000 (Guthrie and Susi, 1963).
Saugstad (1975) determined the incidence and distribution of PKU in Norway. Between 1951 and 1972, the observed incidence of PKU was 0.07 ± 0.01 per 1000 live births. In Denmark, the observed incidence was 0.11 ± 0.01 per 1000 gathered from screening data. The observed value was 0.03 ± 0.01 per 1000 screened in Sweden. In Kuwait, Teebi et al. (1987) found 7 cases of PKU among 451 institutionalized mentally retarded persons.
From the increase in parental consanguinity, Romeo et al. (1983) estimated that the incidence of PKU in Italy is between 1 in 15,595 and 1 in 17,815 (according to 2 different formulas), values not greatly different from that derived from screening programs (about 1 in 12,000). DiLella et al. (1986) cited an incidence of 1 per 4,500 in Ireland (the PKU gene has been considered to be Celtic in origin) and 1 per 16,000 in Switzerland with an average incidence of about 1 per 8,000 in U.S. Caucasians.
Prevalence of PKU
There are no composite data on the prevalence of PKU in the United States.
Biochemical Defect in Classical PKU and Other Forms of Hyperphenylalaninemia
The metabolic defect in classical PKU arises from deficient activity of phenylalanine hydroxylase (PAH) (Scriver et al., 1995). This enzyme catalyzes the conversion of phenylalanine to tyrosine as shown in Figure 1. In humans, PAH is predominantly found in the liver, although small amounts of activity are present in kidney. Significantly, this enzyme is not present in brain, even though the nervous system bears the brunt of the deleterious consequences of PKU.
PAH activity is also dependent on the availability of tetrahydrobiopterin (BH 4), which is a necessary cofactor in the hydroxylation reaction, and molecular oxygen. During the hydroxylation reaction, BH 4 is oxidized to quininoid dihydrobiopterin (qBH 2). Because BH 4 is consumed at stochiometric amounts equal to phenylalanine in the reaction and is in limited supply, the qBH 2 product must be reduced back to the tetrahydro form of biopterin for phenylalanine hydroxylation to proceed efficiently. The reduction of qBH 2 is catalyzed by dihydropteridine reductase (DHPR). Moreover, BH 4 is synthesized de novo by the sequential action of other enzymes, including GTP-cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase and other enzymes that are yet to be well characterized. It is important to note that BH 4 is a cofactor for two other hydroxylase enzymes, tyrosine hydroxylase and tryptophan hydroxylase, that are necessary for the synthesis of the neurotransmitters dopamine and serotonin, respectively. These neurotransmitters are critically important for synaptic transmission and brain function.
In classical PKU and most cases of non-PKU hyperphenylalaninemia, phenylalanine accumulation results from diminished or absent PAH activity. Less commonly, hyperphenylalaninemia also results from genetic defects in DHPR or enzymes catalyzing BH 4 synthesis, which are also associated with deficient dopamine and serotonin synthesis.
Figure 1. The pathway for phenylalanine metabolism in man and biochemical defects associated with hyperphenylalaninemia.
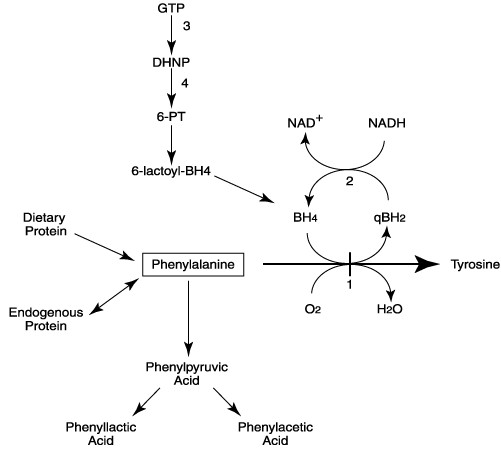 BH4, tetrahydrobiopterin; qBH2, quininoid dihydrobiopterin; GTP, guanosine triphosphate; DHNP, dihydroneopterin triphosphate; 6-PT, 6-pyruvoyl-tetrahydropterin; 6-lacyoyl-BH4, 6-lactoyl tetrahydropterin. Enzymes catalyzing steps are numbered; 1, phenylalanine hydroxylase (PAH); 2, dihydropteridine reductase (DHPR); 3, GTP cyclohydrolase I; 4, 6-pyruvoyl tetrahydropterin synthase.
BH4, tetrahydrobiopterin; qBH2, quininoid dihydrobiopterin; GTP, guanosine triphosphate; DHNP, dihydroneopterin triphosphate; 6-PT, 6-pyruvoyl-tetrahydropterin; 6-lacyoyl-BH4, 6-lactoyl tetrahydropterin. Enzymes catalyzing steps are numbered; 1, phenylalanine hydroxylase (PAH); 2, dihydropteridine reductase (DHPR); 3, GTP cyclohydrolase I; 4, 6-pyruvoyl tetrahydropterin synthase.
In classical PKU, excess phenylalanine is diverted through alternate biochemical pathways that lead to accumulation of phenylpyruvic acid (a phenylketone that originally prompted the name of the disease), phenyllactic acid, phenylacetic acid and other metabolites (Kaufman, 1989) (see Figure 1). The level of these phenylalanine metabolites is directly related to the extent of phenylalanine accumulation. Persons with non-PKU hyperphenylalaninemia having a partial deficiency of PAH typically do not accumulate sufficient phenylalanine to show phenylketonuria.
Phenylalanine is an essential amino acid that cannot be synthesized by humans and must be derived from the diet. In the absence of sufficient dietary phenylalanine, protein synthesis is impaired and the body will catabolize endogenous protein as a source of amino acids. In persons with diminished PAH activity, tyrosine becomes a conditionally essential amino acid, which must also be supplied through dietary sources.
Genetic Basis of Classical PKU and Non-PKU Hyperphenylalaninemia
Genetic causes of hyperphenylalaninemia include primary mutations at the phenylalanine hydroxylase (PAH) gene resulting in classical PKU or non-PKU hyperphenylalaninemia, and defects in tetrahydrobiopterin (BH 4) synthesis due to dihydropteridine reductase deficiency (DHPR); guanosine triphosphate cyclohydrolase I, 6-pyruvoyl tetrahydropterin synthetase deficiency (Scriver et al., 1995). The most common genetic defects leading to hyperphenylalaninemia in man involve mutations in the PAH gene; defects in the other genes account for less than 5% of the patients detected in newborn screening, and will not be discussed here.
More than 400 mutations in the PAH gene have been described (Scriver and Waters, 1999), and are compiled in the PAH mutation database accessible on the World Wide Web at http:// www.pahdb.mcgill.ca/ . Hyperphenylalaninemia due to PAH deficiency can present with a broad spectrum of phenotypes and a continuum of phenylalanine levels (Mallolas et al., 1999).
A variety of mutations have been found in the PAH gene among persons with classical PKU or hyperphenylalaninemia (Table 7). These include deletions, insertions, missense mutations, splicing defects and nonsense mutations. A complete listing of the specific mutations detected in individuals with classical PKU and hyperphenylalaninemia in the United States is included in Appendix A of this report.
Table 7. Types of Mutations in the PAH Gene in Persons with PKU and Hyperphenylalaninemia.
| Mutation Type | Number of Different Mutations | Percentage |
|---|---|---|
| Missense | 255 | 66% |
| Deletion | 56 | 15 |
| Splicing | 47 | 12 |
| Nonsense | 22 | 6 |
| Insertion | 5 | 1 |
Source: Data were compiled from the PAH Mutation Database Website at http:// www.pahdb.mcgill.ca/
In persons from the United States, 76 different mutations were found in the PAH gene Appendix A ( Table A-I). Within each geographic region of the United States, a large number of PAH mutations were reported to cause classical PKU or hyperphenylalaninemia (Table 8). The relative number of mutations in PAH compared to the number of tested individuals appears to be lower in the Midwest region comprised of Illinois, Michigan and Minnesota. It is unclear, however, whether this low number of different mutations per tested individual is a valid observation or represents ascertainment bias arising from, for example, a few large families with many members.
Table 8. Mutations in the PAH gene identified in 593 persons with PKU and hyperphenylalaninemia from the United States. (A total of 76 different mutations were identified)
aThis should be considered a minimum estimate of the number of different mutations causing PKU or hyperphenylalaninemia, because all mutations may not have been detected.
| Geographic Region of the U.S. | Number of Persons with PKU or Hyperphenyl- alaninemia Tested | Number of Different Mutations Detected in the Geographic Region a |
|---|---|---|
| Southeastern (Georgia only) | 71 | 27 |
| Southeastern (other states) | 52 | 25 |
| Midwestern (IL, MI, MN only) | 228 | 12 |
| Midwestern | 76 | 27 |
| Western | 39 | 27 |
| Northeastern | 127 | 42 |
Outside of the United States, different mutations causing classical PKU or hyperphenylalaninemia are listed in Appendix A ( Table A-II). Some of these data have been excerpted and summarized in Table 9. As noted for the United States, all countries or cities had persons with a number of different mutations.
Table 9. Mutations in the PAH gene identified in persons with PKU and Hyperphenylalaninemia from outside of the United States.
a This should be considered a minimum estimate of the number of different mutations causing PKU or hyperphenylalaninemia, because all mutations may not have been detected.
| Geographic Region of the World | Number of Persons Tested | Number of Different Mutations Detected a |
|---|---|---|
| W. Quebec | 57 | 24 |
| E. Quebec | 47 | 18 |
| Montreal | 37 | 17 |
| Iceland | 31 | 9 |
| Norway | 236 | 33 |
| Sweden | 308 | 16 |
| The Netherlands | 52 | 16 |
| Victoria, Australia | 83 | 23 |
In summary, PKU and hyperphenylalaninemia in the United States and abroad are caused by many different mutations in the PAH gene. The types of mutations represent the breadth of genetic changes observed in other genetic diseases.
Clinical Variability of Classical PKU
Clinical Features of Classical PKU
The symptoms of untreated classical PKU include:
- Mental retardation (reduction of IQ)
- Microcephaly
- Delayed speech
- Agitated behavior
- Pigment dilution with fair skin and hair
- Tooth enamel hypoplasia
- Variations in positions, gait and stance such as a crossed leg sitting position (schneidersitz)
- Epilepsy
- Eczema
- 'Mousy' or musty odor
Other symptoms of PKU include tremors and pyramidal tract signs, increased muscle tone and hyperreflexia with 5% of patients developing spastic paraparesis (Berg, 1994). Extrapyramidal signs such as choreoathetosis have also been described (Thompson et al. 1990). Seizures occur in about 25% of individuals with PKU (Berg, 1994). Berg (1994) also describes vomiting, and poor growth as measured by height and weight in the untreated person. Some other clinical features have been noted at a lower incidence, but it is difficult to ascertain whether their appearance is a direct consequence of PKU itself. An example is a scleroderma-like syndrome (Scriver et al., 1995). As with any other disease, some clinical findings may be associated with PKU by chance alone and may not have any causal relationship. The literature contains little about the variability and incidence/prevalence regarding pigment dilution, tooth abnormalities, or the characteristic "mousy" odor (Berg, 1994; OMIM, 2000). One would expect that PKU would be like other conditions in which the affected person has some, but not all, characteristic signs and symptoms.
Cataracts have been reported to be associated with PKU (Kawashima, et al., 1988) but other studies have not borne this out (Pitt and O'Day, 1991). Other manifestations noted by at least some investigators in addition to those listed above include abnormalities of cerebral white matter, fine motor performance deficit, attention deficits, and impaired "executive function" (Weglage et al., 1997). An increased prevalence of congenital heart disease has been suggested (Verkerk et al., 1991), but is more likely to be found in infants whose mothers had hyperphenylalaninemia as part of the so-called (MPKU) maternal phenylketonuria syndrome rather than in persons with classical PKU without maternal hyperphenylalaninemia (Kesby, 1999). Other notable symptoms such as depression and anxiety, may not actually be caused by PKU but result from the stresses of the disease and its treatment (Levy, 1999; Pietz, et al. 1997). Both treated and untreated persons with PKU show abnormal MRI changes in white matter of the brain (Scriver et al., 1995). The clinical significance is unknown.
The Biochemical Phenotype of PKU
Various phenotypes are discussed in relation to PKU. These include the biochemical phenotype, the enzymatic phenotype, the metabolic phenotype (similar to the previous), developmental phenotype, cognitive phenotype and clinical phenotype. The term "phenotype" is used rather loosely in some published reports. For example, Eisensmith et al. (1996) used pretreatment serum phenylalanine levels as their measurement of clinical phenotype.
The clinical classification of PKU and hyperphenylalaninemia has historically relied on the blood phenylalanine level along with the clinical symptoms. With success in newborn screening and prevention of symptoms due to early therapy, the classification of PKU has placed greater emphasis on the biochemical definition of phenotype. For example, serum phenylalanine levels with unrestricted diets above 1200 mM (20 mg/dl) may be defined as classical PKU; between 600 and 1200 mM as mild PKU, and below 600 mM as non-PKU hyperphenylalaninemia (Weglage, et al., 1997). Scriver, et al (1995) define classical PKU as phenylalanine values above 1000 mM. Guldberg et al., (1998), phenotypically divide cases into four arbitrary categories--classic, moderate and mild PKU and mild hyperphenylalaninemia. Kayaalp et al. (1997) divide phenotypes into three categories: PKU, variant PKU and non-PKU hyperphenylalaninemia. Güttler and Guldberg (1994) consider classical PKU greater than 1200 mM, moderate, 900 to 1200 mM, and mild less than 900 mM. In all of these classification schemes, attempts to relate blood phenylalanine concentration, which is a continuum, to discrete clinical phenotypes may be too artificial and dogmatic. This complicates efforts to extrapolate and compile information regarding phenotypes. Wide variation in blood phenylalanine concentrations within a 24 hour period in those with PKU has been noted (MacDonald et al., 1998), and the blood level of phenylalanine may or may not reflect levels in the brain and the consequent central nervous system damage (Eiken et al. 1996). Furthermore, much of the work regarding clinical descriptions was done before the PKU variants had been elucidated.
Genotype-Phenotype Correlations
The clinical expression of a genetic disease is a composite of a person's genotype and environment. Genetic contributions to the phenotype are complex, consisting of allelic heterogeneity within the PAH gene, the position of a mutation (cis vs. trans), allelic complementation, gene dosage, and by genes at other loci (Eisensmith et al 1996; Guldberg et al 1996; Ledley et al 1986; Scriver and Waters, 1999). For example, genes at other loci can influence phenylalanine transport within the brain and the size and metabolic control of the phenylalanine pool (Scriver and Waters, 1999). This molecular heterogeneity for PKU results in a wide variety of phenotypic heterogeneity (Okano et al. 1991). In PKU, the blood phenylalanine concentration is also greatly influenced by the dietary intake of phenylalanine. It is not surprising then that attempts to correlate genotype at the PAH locus with the clinical, or even biochemical, phenotype in PKU will have limited success.
An individual carries two copies of the PAH gene and most people with PKU possess two different PAH alleles owing to the extensive mutational heterogeneity in this gene. Phenylalanine hydroxylase proteins encoded by different mutant alleles within the same individual gene may interact affecting phenotype in various ways and may also influence the holoenzyme and protein folding (Guldberg et al., 1998; Ledley, 1991). This potential interaction complicates attempts to draw phenotype-genotype correlations based on knowing the genotype alone, since few persons with PKU share the same combination of alleles. Consequently, genotype-phenotype correlations can only be established for a relatively few common genotypes (Güttler et al., 1999).
Despite these considerations, the ability to predict the phenotype from genotypic analysis is desirable for initial diagnosis, genetic counseling, screening, long-term prognosis, and optimal management. For example, Eiken et al (1996) noted that patients homozygous for R408W have difficulty in maintaining recommended blood values while this was not true of certain other mutations such as I65T. Presently, classification and treatment largely occurs on the basis of blood phenylalanine levels in the neonatal period but this parameter can fluctuate and has limitations for prognosis related to clinical course and outcomes (Eiken et al. 1996). Severity of the enzyme phenotype, however, does not necessarily correlate with IQ, and there can be great differences in IQ scores in individuals with the same genotype within and among families. Scriver, et al. (1994) note that in the late or untreated individuals with PKU, the cognitive phenotype or IQ score "does not correlate consistently with mutant genotype and residual enzyme activity, as predicted by mutation analysis."
For a few well described genotypes, phenotypic associations can be established with some confidence (Güttler et al., 1999). Persons carrying two "null" mutations (such as deletions causing frameshifts and nonsense mutations leading to premature termination of translation) have a complete absence of functional PAH enzyme and are more likely to have lower mean IQs than those with "milder" missense mutations (Güttler et al. 1999; Lichter-Konecki et al., 1994). Persons who carry one null mutation are functionally hemizygous for the other allele. Guldberg et al. (1998) noted that disease severity is determined in most cases by the least severe of two PAH mutations, and that two mutations associated with similar severity may have less severe phenotypes together than either of the two alone. Even persons who share the same mutant PAH genotype can have different phenotypes. In this regard, Scriver and Waters (1999) noted that there can be discordance "between the mutant PAH genotype, its predicted effect on enzyme function and the associated metabolic phenotype."
Guldberg et al. (1998) reporting on the European Multicenter Study of PAH deficiency estimated that the observed metabolic phenotype matched the predicted phenotype 79% of the time. They attribute discrepancies to a variety of factors including the arbitrariness of the categories of phenotype not reflecting a continuum, and the way in which individual centers analyzed and classified the data. Particular mutations such as the I65T mutation were frequently represented in the discrepant genotype-phenotype associations.
Enns et al. (1999) studied 133 patients from heterogeneous backgrounds in California and found a lack of correlation between genotype and IQ or between mutation severity and pretreatment phenylalanine levels. They speculated that part of the reason for this is that "the relationship between genotype and biochemical and clinical phenotype is complex." Eisensmith et al. (1996) studying 36 persons in a heterogeneous population in the southeastern US found strong correlations between PAH genotype and pretreatment serum phenylalanine levels (the phenotype used) but noted that there were deviations of phenotypes from those predicted by genotypes.
Eiken et al. (1996) examining genotype-phenotype relationships in 108 patients in Norway found that some genotypes, particularly those with the R261Q and G46S alleles, were associated with phenotypic variation but that genotype information was "able to predict the metabolic phenotype in individuals with PKU." In Sweden, Svensson et al. (1991, 1993) found a correlation between genotype and biochemical phenotype.
Koch et al. (1997) evaluated 72 adults with PKU to investigate genotype and phenotypic parameters such as intellectual ability, academic achievement and mental illness. They found a genotypic correlation to metabolic parameters but not to intellectual abilities. However, some participants were on restricted diets all their lives and others had discontinued the diet at various points in childhood. Trefz et al. (1993) suggested using genotyping to correlate with predicted residual activity of the phenylalanine hydroxylating system. DiSilvestre et al. (1991) noted that although the genotypes of their patients predicted the biochemical phenotype, they did not necessarily predict the clinical phenotype.
Ramus et al. (1999) examined the relationship between PAH genotype and intellectual phenotype in untreated persons with PKU. There were differences in the intellectual phenotypes of persons with the same genotype both within families and in unrelated individuals. They found considerable overlap between their groups and proposed that one or more other genes might be modifiers since the differences could not be explained by dietary differences. Ramus et al. (1993) found that patients in one family with a genotype predictive of almost no enzyme activity were not severely retarded.
Mallolas et al. (1999) studied the genotype-phenotype correlation in 115 patients in Catalonia and their relatives. They classified patients into 3 groups--PKU, variant PKU and non-PKU hyperphenylalaninemia based on phenylalanine levels at diagnosis and "the evolution of the patient" (not explained). Certain mutations correlated with these categories and predicted residual activity as expected, but in four cases the phenotype was not consistent with the prediction. Okano et al. (1998) divided clinical phenotypes into classical PKU and non-classical PKU and determined that genotypic predictions were useful. Kayaalp et al. (1997) in an analysis of 31 reports concluded that there were broad correlations between mutant PAH genotypes and phenotypes, and that the majority of PAH mutations conferred a consistent phenotype but that discordances did exist. They noted that when examining clinical phenotype, IQ itself is a complex trait and that hyperphenylalaninemia itself sometimes behaves like a complex trait as well. They identified 11 mutations with an inconsistent phenotypic correlation.
Clinical Variation of PKU within Sibships
An estimate of clinical variability might be obtained by comparing siblings with PKU who share the same genotype at the PAH locus, have 50% of their other genes in common, and have a similar family environment. Nevertheless, surprisingly few reports appear in the literature describing clinical variability in sibs with PKU. Two children who were siblings discordant as to sex had the identical genotype at the PAH locus, and had identical hydroxylation rates but different renal excretion rates of phenylalanine transamination metabolites and protein accretion (Treacy et al. 1996). This report did not describe clinical characteristics. Two untreated sibs with PKU who had identical PAH genotypes had differences in intellectual functions. One was mildly retarded while the other was severely retarded (Özalp, 1994). Pérez et al. (1994) describe two Spanish families with PKU. In one, the plasma phenylalanine levels in two PAH deficient sibs were different after a standard protein load. In the second family, the untreated phenylalanine levels were the same but the cognitive development of the sibs differed. Further details were not provided. Within families, variation in intellectual outcome of untreated persons with the same genotype has been described (Ramus et al. 1993). Tyfield, et al. (1990) described a family in which two first cousins had different biochemical presentations in infancy but because of the rare, private mutation they had, DNA analysis could not explain the different biochemical phenotypes. In a Romanian family, two sibs had identical PAH genotypes, however one had been diagnosed as having non-PKU hyperphenylalaninemia, while the other had been diagnosed with classic PKU based on blood phenylalanine levels. Their clinical status was not compared (Popescu et al. 1994). Ramus et al. (1999) described 12 pairs of siblings with the same genotype who had variation in their intellectual phenotype that was often considerable. Verkerk and van der Meer (1995) report a case of fraternal twins in which the female twin initially had a false negative Guthrie test, but who was retested because her brother had been detected with elevated plasma phe, thus showing that plasma phenylalanine levels can vary at different times even within a twin pair. Hsiao et al. (1986) described two sibs with PKU both of whom had similar IQs of 69 and 75. In one family described by Guldberg, Levy, Koch et al. (1994), discordant phenotypes within the family were ascribed to mutations associated with both PKU and hyperphenylalaninemia.
Clinical Variation in Neurological Findings
Demyelination may be seen in untreated adults with low brain weight and reduction in total white matter. Diffuse cortical atrophy and reduced dendritic arborization have been described ( Pietz, 1998; Baumann and Kemper, 1982). Spasticity, irritability and other symptoms occur in untreated PKU and may be seen after cessation of treatment (McCombe et al. 1992; Wood, 1976). An increased incidence of neurological signs such as tremor, fine motor skill reduction, brisk deep tendon reflexes and clumsy motor coordination have been described in adult individuals with PKU not on dietary phenylalanine restriction (Pietz, Dunckelmann et al., 1998). Some of these signs were subtle, but were a sensitive marker of diffuse brain impairment.
White matter abnormalities have been described in some individuals with PKU (Alvord et al., 1950; Battistini et al., 1991; Cleary et al., 1994; Cleary et al., 1995; McCombe et al., 1992; Poser and Van Bogaert, 1959; Shaw et al., 1991; Shaw et al., 1990; Thompson et al., 1990; 1991). The clinical significance of the white matter changes is not clear to many researchers and clinical severity may not parallel imaging severity (Cleary et al., 1994; Leuzzi et al., 1993; Pearsen et al., 1990). White matter disease seen on MRI does not often correlate with clinical parameters such as IQ, neurological deficits, or fine motor abilities (Ullrich et al. 1994). Furthermore, at least some reversal of MRI changes in cerebral white matter occurs when the blood phenylalanine is lowered by dietary changes (Battistini et al., 1991; Cleary et al., 1995; Cleary et al., 1994; Weglage et al., 1993). Weglage et al. (1993) described two cases with no alterations in white matter and one with changes that regressed with dietary controls. Battistini and colleagues (1991) described an 18-year-old female who was placed on dietary restriction immediately after birth until 5 years of age and who had normal psychomotor development and school performance until demonstrating anxiety and depression. The neurologic examination revealed generalized hypotonia and weak abdominal reflexes, and the MRI showed abnormalities as did the auditory evoked potentials and visual evoked potentials. These changes reversed after two years on a low phenylalanine diet. Bick and colleagues (1993) reported that white matter abnormalities in persons with treated hyperphenylalaninemia represented reversible structural myelin changes rather than permanent demyelination. Normal MRIs were found only in patients with plasma phenylalanine levels continually below 360 µmol/L in the study by Bick and colleagues (1993). Leuzzi, Gualdi et al., 1993 and Leuzzi, Trasimeni et al. (1995) suggested that the white matter abnormalities found in their studies of children and adults were related to recent sustained exposure to high phenylalanine concentrations. Others have found that individuals with PKU with adequate dietary control had less severe white matter abnormalities than those who were poorly controlled (Shaw et al., 1991). Weglage et al. (1997) speculated that there is an individual vulnerability of the brain to elevated phenylalanine levels in persons with PKU, a view shared by Ullrich et al. (1994) who use as evidence that normal MRI findings may be seen in untreated individuals with PKU. Ullrich et al. (1994) also postulated that there might be different susceptibilities in the brain to phenylalanine at different ages. Toft et al. (1994) found less frequent white matter changes in children with PKU than in adolescents, suggesting that the myelin abnormalities progress with time.
Electrophysiologic studies in PKU have indicated abnormal nerve conduction in some patients. Visual evoked potential abnormalities have been found in some cases of PKU (Ludolph et al., 1996; McCombe et al. 1992) but not in others (Villasana et al., 1989). Visual evoked potential changes and white matter abnormalities found by MRI correlate in some studies (Bick et al. 1990 in Battistini et al., 1991) but not others (Bick et al., 1991; 1993; Cleary et al., 1994). Consistent with those who found delayed visual evoked potentials, visual event-related potentials (ERPs) as measured by Henderson et al. (2000), found delayed early ERPs. They suggested that this is due to a myelin defect or low dopamine levels perhaps due to high phe levels that interfere with dopamine production. Similarly, abnormalities of auditory evoked potentials in early treated individuals with PKU were described by some investigators (Cardona et al., 1991; Coskun et al., 1993), but not by others (Ludolph et al., 1996).
Abnormal EEG findings of generalized paroxysmal activity and general slowing that worsens with advancing age have been described in PKU (Pietz et al., 1988; Gross, et al. 1981). Blascovics et al. (1981) found similar EEG changes, but these had normalized by 6 years of age in almost all of their patients.
Cerebral atrophy in PKU has been reported rarely (Behbehani et al., 1981; Leuzzi, Gualdi et al., 1993; Leuzzi et al., 1995; Mostafawy et al., 1970; Pearsen et al., 1990). Brain calcification was reported in one case (Kawashima et al. 1988).
Variation in Mental Retardation
Mental retardation is considered the major manifestation of untreated PKU, and the vast majority of individuals with PKU are profoundly or moderately retarded (Scriver et al., 1995). Yet there have been persons with classical PKU who were not mentally retarded. The cognitive phenotype may vary even when the metabolic phenotype is similar. Untreated subjects with the same genotype can show large variation in the intellectual outcome (Tyfield, 1997), and some of this may be due to variation in phenylalanine flux through other pathways involved in phenylalanine homeostasis (Treacy et al., 1993). In one report, a Bedouin woman was diagnosed with PKU after she had 3 children with maternal PKU. She was of low normal intelligence but not mentally retarded (Usha et al., 1992). Another report by Pitt (1971) described two cases of PKU with normal intelligence. Primrose (1983) described PKU with normal intelligence. In another report, Tyfield et al. (1995) describe two older men in a family where two cousins had PKU diagnosed in infancy who both had "moderately raised blood phenylalanine concentrations" and showed no signs of impaired intelligence. No information on the type of disorder or levels is provided. Ramus et al. (1999) reported on genotyping and intellectual phenotype and noted some persons with PKU who had normal intelligence despite not being treated. Trefz et al. (1993) described a woman, with a normal IQ on an unrestricted diet, who was diagnosed with PKU at 23 years of age after giving birth to a child with signs defects secondary to maternal PKU. Özalp et al. (1994) reported two patients homozygous for the R261Q mutation who were profoundly retarded but two untreated sisters with the same genotype were reported as intellectually normal. In a large population survey of randomly selected adults in Ontario, Canada, two men were found, one with classic PKU and the other with persistent benign hyperphenylalaninemia. Both had normal intelligence (Advisory Committee on Inborn Errors of Metabolism to the Ontario Ministry of Health, 1976). Eiken et al. (1996) noted that in two untreated patients who were homozygous for the G465 mutation, one was severely mentally retarded while the second was mildly affected. Hsiao et al. (1986) described a family in which the 16-year-old male had mild mental retardation while one of his elder sisters had classic PKU and an IQ of 75. DiSilvestre and colleagues (1991) described three siblings with identical PAH genes who had different clinical manifestations and different intellectual outcomes. One had a normal IQ, one was severely retarded and one was on a phenylalanine restricted diet until 6 years of age and now as an adult, has a normal IQ. Hanley et al. (1999) described 6 women who were in the range of normal intelligence who were diagnosed with PKU after giving birth to one or more affected offspring while another 8 were found during prenatal screening activities. Perry et al. (1973) described four siblings, three of whom had normal intelligence who were diagnosed as adults. Superti-Furga et al (1991) reported on two sisters with PKU who had no mental retardation who each gave birth to a child with signs of maternal PKU syndrome. Hanley et al. (1999) reviewed the literature and found, in all, 23 reports of women with previously untreated PKU with IQs that were normal or close to normal who were diagnosed after they had affected offspring. They suggest that between 2 to 3% and 20% of all subjects with untreated PKU may "have intelligence within or near the reference range." These studies show that even within the hallmark sign of PKU, mental retardation, clinical variability exists, and mental retardation does not invariably occur even with elevated blood phenylalanine and a classical PKU genotype.
Variation in Neuropsychiatric Deficits
A variety of neuropsychiatric and psychological deficits have been identified by various researchers in PKU. Early studies in this area often included subjects with non-PKU hyperphenylalaninemia, measured phe levels in various ways and points in time, or used varying definitions of PKU. In this section, only a brief review of a selected sampling of studies are included. Many of these studies have been hampered by lack of control groups, inconsistency of measures chosen across studies making comparison difficult, use of subjects on diet who may not have verified phenylalanine levels, comparison of subjects off diet for varying length of time, failure to account for phenylalanine levels of subjects at testing, and small numbers of subjects studied. It is also difficult to ascertain which parameters are the specific result of PKU and which are secondary to the effects of having a chronic illness. Comparisons on and off diet are not included in this brief review. Burgard and colleagues (1994) examined 40 symptoms related to emotional, antisocial and conduct disorders as well as specific symptoms such as tics and eating disorders. While the frequency of occurrence was higher in the adolescents with PKU who were studied, the conclusion was that the disturbances arose more from stress in chronic illness rather than the PKU itself.
Weglage and colleagues (1996) concluded that even those children with PKU who had normal IQs and were treated early had impaired attentional control mechanisms. They found some correlation of impaired attention with elevated phenylalanine levels and speculated that impairment of frontal lobe function was a result of phenylalanine dependent dopamine deficiency. A declining IQ may be seen when phenylalanine levels are elevated over time (Burgard et al. 1996).
Other neuropsychiatric traits such as concentration problems, restlessness, slower reaction times and reduced information processing abilities have been reported in those who have high serum phenylalanine levels after diet discontinuation which might also affect IQ and school performance (Weglage et al., 1992). Reduced attentional abilities, slow information processing and slow motor reaction time have also been identified (Schmidt et al., 1994). Information processing in a study by Stemerdink et al. (1995) did not show impairment suggesting clinical variation in this parameter.
Pennington and Smith (1983) suggest that children with PKU even when treated showed certain deficits including those of flexible thinking, conceptual skill, tests of right hemisphere spatial abilities as well as problems in mathematics, attention, and organizational and planning skills. They propose a hypothesis of prefrontal dysfunction. Executive function deficits were also found by Welsh et al. (1990) and by Diamond et al. (1997) but not by Mazzocco et al. (1992 in Burgard et al., 1999).
Agoraphobia (an irrational fear of leaving the setting of home) has been described after treatment has been discontinued in young adult individuals with PKU (Waisbren & Levy, 1991).
Delayed speech development in children with PKU has been described by Melnick et al. (1981) in some of their study participants, but this was not found by Ozanne et al. (1990). Linguistic development was found to be delayed in some study participants by Melnick et al. (1981) and Berry and colleagues (1979) but not by Zartler and Sasserman (1981). Visual perception and visual motor deficits have been identified when children with PKU were compared to their healthy siblings (Fishler et al., 1989), but no differences were found in language and perceptual skills. Michel et al. (1990) reported deviations from norms in visual perception at 5 years of age, but not one year later. This group did not find any abnormalities in motor or language development.
Burgard et al. (1999) reviewed many of the neuropsychological results for such functions as higher cognitive skills, memory, executive functions, simple reaction time, and sustained attention, citing evidence from different studies showing impaired and intact function for the same parameters.
Clinical Variation Associated with Brain Phenylalanine Concentration
Recent studies utilizing proton magnetic resonance spectroscopy (MRS) suggests that the phenylalanine concentration in the brain may be critically important for understanding the basis for clinical variation in PKU. Using this noninvasive method, elevated levels of phenylalanine have been detected in the brains of persons with PKU (Novotny et al., 1995; Kreis et al., 1995; Pietz et al., 1995; Möller et al., 1998). Although MRS lacks the sensitivity to detect minor phenylalanine metabolites, most studies have found no abnormalities in the levels of other major brain metabolites that are markers of neuronal metabolism. Möller et al (1998) investigated 11 individuals with PKU with MRS and showed that brain phenylalanine concentration rose when patients were given an oral phenylalanine load. Three atypical untreated women with PKU normal IQs had elevated blood phenylalanine but normal brain phenylalanine concentrations. When challenged with the oral phenylalanine load, they showed a blunted rise in brain phenylalanine compared to more typical individuals with PKU. These results suggest that the PKU phenotype may be more closely associated with the phenylalanine concentration in brain than in the blood. Phenylalanine elevations in the brain may cause biochemical changes such as impaired production of dopamine and serotonin, which in turn produce clinical effects (Krause et al., 1985).
The most common skin lesion in PKU is a scaling eczematous dermatitis seen in about 25% of all patients (Berg, 1994). Symptoms of eczema are probably due to the toxic effects of phenylalanine and its products such as phenylpyruvic acid on the skin (Jablonska et al., 1967; Nova et al., 1992). Pigment dilution resulting in skin, hair and irises of the eyes that are paler than their parents is thought to be due to decreased melanin and DOPA production (Nova et al., 1992) seen in PKU. There have been consistent reports of scleroderma-like lesions and these are considered by Nova et al. (1992) to be one of the major types of cutaneous lesions seen associated with PKU. The sclerodermatous lesions showing skin and muscle indurations have been noted by a number of researchers (Brown et al., 1986; Coskun et al., 1990; Drummond et al., 1966; Jablonska et al., 1967; Kornreich et al., 1968; Lasser et al., 1978; Nishimura et al., 1959; Nova et al., 1992). The lesions have been reported as most noticeable in the arms and buttocks, and dimpling often occurred initially during the first two years of life (Jablonska et al. 1967; Nova et al., 1992). Improvement of the lesions with dietary control was noted in some patients, implying a causal relationship with PKU (Coskun et al., 1990).
Bone Mineral Density
A decrease in total bone mineral density and spine bone mineral density as measured by dual-energy x-ray absorptiometry was found in 32 prepubertal children with PKU on an adequate diet as compared to 95 age-matched controls (Allen et al., 1994). This confirms previous studies in adults using radiologic methods (Holt and Allen, 1967; Woodring and Rosenbaum, 1981) and CT scans (Carson et al., 1990). A previous study in children indicated a normal bone mineral density but a lower bone mineral content (McMurry et al., 1992). A potential but unproven consequence of altered bone mineral density could be an increased propensity to fractures.
Pathogenesis of PKU
It is likely that no single pathogenic mechanism is responsible for the brain abnormalities in PKU. The central biochemical abnormality in PKU is an elevation in the phenylalanine concentration in tissues, which results in secondary accumulation of several phenylalanine metabolites. There seems to be consensus, however, that the primary "toxic" agent in PKU is phenylalanine itself (Scriver et al., 1995). Although various phenylalanine metabolites (e.g., phenylpyruvate, phenylacetate) are known to have deleterious effects on a variety of metabolic pathways (Patel and Arinze, 1975), these compounds do not accumulate to high enough levels in individuals with PKU to have an impact on brain metabolism (Kaufman, 1989).
There is little support for the hypothesis implicating tyrosine deficiency as primarily responsible for neurologic symptoms in PKU (Scriver et al., 1995). Untreated individuals with PKU show no consistent reduction in plasma tyrosine concentration (Koepp and Held, 1977) and dietary tyrosine supplementation alone without phenylalanine restriction does not prevent the mental retardation of PKU (Batshaw et al., 1981).
The most striking pathologic abnormality in PKU is a deficiency of brain white matter, or myelin. Using sensitive MRI techniques, the incidence of abnormal white matter in PKU patients is greater than 90% in most studies. How does the myelin abnormality originate in PKU? Using the PKU Pah enu2 mouse, Dyer and colleagues (1996) showed that the oligodendrocytes that are responsible for myelin synthesis remained viable in the brain but failed to produce myelin in normal quantities. Oligodendrocyte cultures derived from PKU mice were capable of differentiating normally in vitro when grown under low phenylalanine conditions (2 mg/dl) and synthesized myelin proteins and myelin-specific galactocerebroside lipid. However, oligodendrocyte cells grown with high phenylalanine (10-40 mg/dl) showed an increased amount of glial fibrillary acidic protein and had an aberrant cytosolic localization of myelin/oligodendrocyte-specific protein, which is normally present in the plasma membrane (Dyer et al., 1996). These results suggest that the oligodendrocytes fail to synthesize myelin components at a normal rate under conditions of hyperphenylalaninemia.
At the biochemical level, Hommes (1991) has hypothesized that the key step leading to impaired myelin synthesis is inhibition of oligodendrocyte-specific ATP-sulfurylase by phenylalanine. This enzyme plays an important regulatory role in the utilization of sulfate in the brain and is necessary for the synthesis of sulfatide lipids, which are prominent in normal myelin and deficient in PKU myelin. ATP-sulfurylase is inhibited by phenylalanine (Hommes and Matsuo, 1988). In rats made hyperphenylalaninemic by administration of phenylalanine together with PAH inhibitors (Berger et al., 1980; Hommes et al., 1982) and in the PAH-deficient mouse (Hommes and Moss, 1992), brain myelin shows an increased rate of turnover. The combination of increased myelin turnover with impaired myelin synthesis would lead to myelin deficiency.
In addition to its overall deficiency in the brain, myelin lipids from experimental PKU animals (Johnson and Shah, 1973) and humans with PKU (Shah, 1979) have an abnormal fatty acid composition with reductions in specific unsaturated fatty acids report also indicates that glucose utilization in the abnormal white matter of individuals with PKU is impaired compared to normal controls (Hasselbalch et al., 1996), but this may be secondary to abnormal myelin and not specific to PKU.
In contrast to myelin deficiency, the pathogenesis of mental retardation and neuropsychological test abnormalities in PKU is even less firmly established. Hyperphenylalaninemia has been found to have a variety of deleterious effects on neuronal metabolism, including:
- Inhibition of amino acid transport into the brain. Phenylalanine is transported into the brain across the blood-brain barrier by a transporter system that is specific for large neutral amino acids, including tyrosine, tryptophan, methionine, histidine and the branched chain amino acids (Pardridge, 1977). In PKU, elevated phenylalanine competitively inhibits the influx of these amino acids across the blood-brain barrier. Moreover, the transport of tyrosine (Aragon et al., 1982) and tryptophan (Herrero et al., 1983) into synaptosomal plasma membrane vesicles is inhibited by phenylalanine. This inhibition by phenylalanine of amino acid transport within the brain may result in a relative deficiency of certain critical amino acids and cause secondary detrimental effects on brain metabolism.
- Inhibition of brain protein synthesis. Protein synthesis in the brain is inhibited in direct proportion to the degree of hyperphenylalaninemia (Wall and Pardridge, 1990). The mechanism involves polyribosome disaggregation (Binek et al., 1981), inhibition of initiation of translation (Elsliger et al., 1989) and impaired polypeptide elongation (Binek et al 1981, Binek-Singer and Johnson, 1982). Deficiency of large neutral amino acids in the brain may be responsible for the protein synthesis defect, because the effect of phenylalanine may be reversed by supplementation with a mixture of these neutral amino acids (Binek-Singer and Johnson, 1982). It seems reasonable to conclude that chronic inhibition of brain protein synthesis would contribute in part to the poor brain growth seen in rats made hyperphenylalaninemic (Huether et al., 1983; Burri et al., 1990) and persons with untreated PKU.
- Inhibition of synthesis of dopamine and serotonin. There is an inverse correlation between plasma phenylalanine concentration and synthesis of the neurotransmitters dopamine and serotonin in the brains of individuals with PKU (Güttler and Lou, 1986). Two mechanisms probably account for reduced neurotransmitter synthesis. One mechanism may involve diminished brain transport and availability of tyrosine and tryptophan, which are two amino acids precursor substrates for the synthesis of dopamine and serotonin, respectively as noted above. Evidence also suggests that phenylalanine directly inhibits neurotransmitter synthetic enzymes (Grahame-Smith and Moloney, 1965; Ikeda et al., 1967). This neurotransmitter deficiency may underlie some of the neuropsychologic and behavioral abnormalities in PKU. However, it is unclear whether neurotransmitter deficiency alone is insufficient to account for the cognitive dysfunction in PKU, since treatment of individuals with PKU with L-dopa, a precursor to dopamine that bypasses the brain tyrosine deficiency, did not lead to an improvement on neuropsychologic tests or visual-evoked potentials (Ullrich et al.,1996).
- Decrease in muscarinic acetylcholine receptors and high voltage calcium channels. In rats made hyperphenylalaninemic, muscarinic acetylcholine binding sites in the cerebrum of the brain showed a gradual reduction over time (Matsuo and Hommes, 1988). After 21 days, the reduction in acetylcholine receptors became persistent and did not return to normal with reversal of the hyperphenylalaninemia. A 13-37% reduction of muscarinic acetylcholine receptors was also demonstrated in the hippocampus of the HPH-5 mouse brain during sustained hyperphenylalaninemia (Hommes, 1994). The acetylcholine receptors in the frontal, parietal and occipital lobes showed a more modest reduction, whereas the number of receptors in the putamen was unchanged. In cultured hippocampal cells derived from newborn rats made hyperphenylalaninemic, the amplitude of high voltage-activated calcium channels were down-regulated, whereas low voltage channels were not affected (Martynyuk et al., 1991). This effect seems to be mediated at the mRNA level (Dzhura et al., 1998). These initial studies suggest that critical membrane functions in the neurons are disturbed in hyperphenylalaninemia, leading to speculation that defects in neuronal signaling may be important in the pathogenesis of the mental retardation of PKU.
It is important to interject a word of caution in interpreting many of the animal studies published to date. Most studies have utilized an animal model for hyperphenylalaninemia that relies on the administration of phenylalanine together with an inhibitor of PAH, usually alpha-methylphenylalanine or p-chlorophenylalanine. These inhibitors probably have additional metabolic effects that may be unrelated to the phenylalanine pathway. Some studies have incorporated proper controls that should have detected extraneous effects of the inhibitors, i.e. animals treated with inhibitor alone without phenylalanine supplementation. The recent availability of PAH-deficient mouse models (i.e. HPH-5 and Pah enu-2 mice) should provide a more appropriate model for PKU (McDonald, 1994), but relatively few studies have been done on these mice so far.
References
- Advisory Committee on Inborn Errors of Metabolism to the Ontario Ministry of Health. Phenylketonuria variants in Ontario. CMA Journal 1976;115:509-512.
- Allen JR, Humphries IRJ, Waters DL, et al. Decreased bone mineral density in children with phenylketonuria. American Journal of Clinical Nutrition 1994;59:419-22
- Alvord E, Stevenson L, Vogel R, Angle R. Neuropathological findings in phenyllpyruvic oligophrenia (phenylketonuria). J Neuropathol Exp Neurol 1950; 9:298-310.
- American Academy of Pediatrics, Committee on Genetics, Newborn Screening Fact Sheets (RE9632), Pediatrics 1996;Vol 98 (3) September; 473-501.
- Aragon MC, Gimenez C, Valdeveso F. Inhibition by L-phenylalanine of tyrosine transport by synaptosomal plasma membrane vesicles: Implication in the pathogenesis of phenylketonuria. J Neurochem 1982;39:1185.
- Batshaw ML, Valle D, Bessman SP. Unsuccessful treatment of phenylketonuria with tyrosine. J Pediatr 1981;99:159-160.
- Battistini S, De Stefano N, Parlanti S, Federico A. Unexpected white matter changes in an early treated PKU case and improvement after dietary treatment. Funct Neurol 1991;6:177-180.
- Bauman ML, Kemper TL. Morphologic and histoanatomic observations of the brain in untreated human phenylketonuria. Acta Neuropathol (Berl) 1982;58(1):55-63.
- Behbehani, AW, Krtsch H, Schulte FJ. Cranial computerized tomography in phenylketonuria. Neuropediatrics 1981;12:295-302.
- Berg BO. Child neurology: a clinical manual. 2nd ed. 1994; Philadelphia, JB Lippincott.
- Berry HK, O'Grady DJ, Perlmutter LJ, Bofinger MK. Intellectual development and academic achievement of children treated early for phenylketonuria. Dev Med Child Neurol. 1979;21:311-320.
- Bick U, Fahrendorf G, Ludolph A, Kurt U. Alterations of myelin in treated patients with hyperphenylalaninemia (HPA). In: Proceedings of the Vth International Congress on Inborn Errors of Metabolism, Asilomar, Pacific Grove, Ca, June 1-3, 1990, abstract W4.9. In Battistini et al., 1991.
- Bick U, Fahrendorf G, Ludolph AC, et al. Disturbed myelination in patients with treated hyperphenylalaninemia: evaluation with magnetic resonance imaging. Eur J Pediatr 1991;150:185-9.
- Bick U, Ullrich K, Stöber U, et al. White matter abnormalities in patients with treated hyperphenylalaninemia: magnetic resonance relaxometry and proton spectroscopy findings. Eur J Pediatr 1993:152:1012-20.
- Binek PA, Johnson TC, Kelley CJ. Effect of methylphenylalanine and phenylalanine on brain polyribosomes and protein synthesis. J Neurochem 1981;36:1476-84.
- Binek-Singer P, Johnson TC. The effects of chronic hyperphenylalaninaemia on mouse brain protein synthesis can be prevented by other amino acids. Biochem J 1982;206:407-414.
- Blascovics ME, Engel R, Podosin R, et al. EEG patterns in phenylketonuria under early initiated dietary treatment. Am J Dis Child. 1981;135:802-8.
- Brown EH, Berry HK, Olson J, Levinson J. Phenylketonuria and scleroderma. J Inher Metab Dis. 1986;9:405-6.
- Burgard P, Armbruster M, Schmidt E, Rupp A. Psychopathology of patients treated early for phenylketonuria: results of the German collaborative study of phenylketonuria. Acta Paediatr Suppl 1994;407:108-10.
- Burgard, P., Bremer, H.J., Bührdel P, et al. Rationale for the German recommendations for phenylalanine level control in phenylketonuria 1997. European Journal of Pediatrics,1999; 158, 46-54.
- Burgard P, Schmidt E, Rupp A, et al. Intellectual development of the patients of the German Collaborative Study of children treated for phenylketonuria. Eur J Pediatr. 1996;155 Suppl 1:S33-8.
- Berger R, Springer J, Hommes FA. Brain protein and myelin metabolism in young hyperphenylalaninemic rats. Mol Cell Biol 1980;26:31-36.
- Burri R, Matthieu J-M, Vandevelde M, et al. Brain damage and recovery in hyperphenylalaninemic rats. Dev Neurosci 1990;12:116-25.
- Cardona F, Leuzzi V, Antonozzi I, Benedetti P, Loizzo A. The development of auditory and visual evoked potentials in early treated phenylketonuric children. Electroencephalogr Clin Neurophysiol 1991 Jan-Feb;80(1):8-15.
- Carson DJ, Greeves LG, Sweeney LE, Crone MO. Osteopenia and phenylketonuria. Pediatr Radiol 1990; 20:598-599.
- Carter CO, Woolf LI. The birthplaces of parents and grandparents of a series of patients with phenylketonuria in southeast England. Ann Hum Genet 1961;25:57-64.
- Centerwall WR, Neff CA. Phenylketonuria: a case report of children of Jewish ancestry. Arch Pediatr 1961;78:379-384.
- Cleary, MA, Walter JH, Wraith JF, et al. Magnetic resonance imaging of the brain in phenylketonuria. Lancet 1994;344:87-90.
- Cleary, MA, Walter JH, Wraith JF, et al. Magnetic resonance imaging in phenylketonuria: reversal of cerebral white matter change. J Pediatr 1995;127:251-5.
- Cohen BE, Bodonyi E, Szeinberg A. Phenylketonuria in Jews. Lancet 1961;I:344-345.
- Coskun T. Ozalp I, Kale G, Gogus S. Scleroderma-like skin lesions in
- two patients with phenylketonuria. Eur J Pediatr 1990;150:109-10.
- Coskun, T, Topcu M, Üstündag I, et al. Neurophysiological studies of patients with classical phenylketonuria: evaluation of results of IQ scores, EEG and evoked potentials. Turk J Pediatr 1993;35:1-10. In Ludolph et al. 1996.
- Diamond A., Prevor MB, Callender G, Druin DP. Prefrontal cortex cognitive deficits in children treated early and continuously for PKU. Monogr Soc Res Child Dev. 1997;62:1-208.
- DiLella AG, Kwok SCM, Ledley FD, et al. Molecular structure and polymorphic map of the human phenylalanine hydroxylase gene. Biochemistry 1986; 25:743-749.
- DiSilvestre D, Koch R, Groffen J. Different clinical manifestations of hyperphenylalaninemia in three siblings with identical phenylalanine hydroxylase genes. Am J Hum Genet. 1991;48:1014-6.
- Drummond KN, Michael AF, Good RA. Tryptophan metabolism in a patient with phenylketonuria and scleroderma. Can Med Assoc J. 1966;94:834-8.
- Dyer CA, Kendler A, Philibotte T et al. Evidence for central nervous system glial cell plasticity in phenylketonuria. J Neuropath Exp Neurol 1996;55:795-814.
- Dzhura I, Naidenov, V, Zhuravleva S, et al. Expression of Ca 2+ channels from rat brain with model phenylketonuria in Xenopus oocytes. Brain Res 1998; 783:280-85.
- Eiken HG, Knappskog PM, Motzfeldt K, et al. Phenylketonuria genotypes correlated to metabolic phenotype groups in Norway. Eur J Pediatr. 1996;155:554-60.
- Eisensmith RC, Martinez DR, Kuzmin AI, et al. Molecular basis of phenylketonuria and a correlation between genotype and phenotype in a heterogeneous southeastern US population. Pediatrics 1996;97:512-6.
- Elsliger M-A, Thériault GR, Gauthier D. In vitro localization of the protein synthesis defect associated with experimental phenylketonuria. Neurochem Res 1989; 14:81-84.
- Enns GM, Martinez DR, Kuzmin AI, et al. Molecular correlations in phenylketonuria: mutation patterns and corresponding biochemical and clinical phenotypes in a heterogenous California population. Pediatric Research. 1999;46:594-602.
- Fishler K, Azen CG, Friedman EG, Koch R. School achievement in treated PKU children. J Ment Defic Res. 1989. 33:493-8.
- Grahame-Smith DH, Moloney L. The subcellular localization, purification and properties of tryptophan 5-hydroxylase in brain. Biochem J 1965;96:66P.
- Gross PT, Berlow S, Schuett VE, Fariello RG. EEG in phenylketonuria. Attempt to establish clinical importance of EEG changes. Arch Neurol.1981;38:122-6.
- Guldberg P, Levy HL, Henriksen KF, Güttler F. Three prevalent mutations in a patient with phenylalanine hydroxylase deficiency: implications for diagnosis and genetic counselling. J Med Genet. 1996;3:161-4.
- Guldberg P, Levy HL, Koch R, et al. Mutation analysis in families with discordant phenotypes of phenylalanine hydroxylase deficiency. Inheritance and expression of the hyperphenylalaninemias. J Inher Metab Dis 1994;17:645-51.
- Guldberg, P, Rey F, Zschocke J, et al. A European Multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. American Journal of Hum Genet 1998;63:71-79.
- Guthrie R, Susi A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 1963;32:338-343.
- Güttler F, Guldberg P. Mutations in the phenylalanine hydroxylase gene: genetic determinants for the phenotypic variability of hyperphenylalaninemia. Acta Paediatr Suppl 1994; 407:49-56.
- Güttler F, Guldberg P, Eisensmith RC, Woo, SLC. Molecular genetics and outcome in PKU. Mental Retardation and Developmental Disabilities Research Reviews. 1999;5:113-6.
- Güttler F, Lou H. Dietary problems of phenylketonuria: effect on CNS transmitters and their possible role in behavior and neuropsychological function. J Inher Metab Dis 1986;9:169-77.
- Hanley WB, Platt LD, Bachman RP, et al. Undiagnosed maternal phenylketonuria: the need for prenatal selective screening or case finding. Am J Obstet Gynecol. 1999;180:986-94.
- Hasselbalch S, Knudsen GM, Toft PB et al. Cerebral glucose metabolism is decreased in white matter changes in patients with phenylketonuria. Pediatr Res 1996; 40:21-24.
- Henderson RM, McCulloch DL, Herbert AM, Robinson PH. Visual event-related potentials in children with phenylketonuria. Acta Paediatr 2000;89:52-7.
- Herrero E, Aragon MC, Gimenez C, Valdiveso F. Inhibition by L-phenylalanine of tryptophan transport by synaptosomal plasma membrane vesicles: Implications in the pathogenesis of phenylketonuria. J Inher Metab Dis 1983;6:32.
- Hofman KJ, Steel G, Kazazian HH, Valle D. Phenylketonuria in U.S. blacks: molecular analysis of the phenylalanine hydroxylase gene. Am J Hum Genet 1991;48:791-798.
- Holt JF, Allen RJ. Radiologic signs in the primary aminoacidurias. Ann Radiol 1967;10:317-21.
- Hommes FA. On the mechanism of permanent brain dysfunction in hyperphenylalaninemia. Biochem Med Metab Biol 1991; 46:277-87.
- Hommes FA. Loss of neurotransmitter receptors by hyperphenylalaninemia in the HPH-5 mouse brain. Acta Paediatr Supple 1994; 407:120-1.
- Hommes FA, Eller AG, Taylor EH. Turnover of the fast component of myelin and myelin protein in experimental hyperphenylalaninemia. Relevance to termination of dietary treatment in human PKU. J Inher Metab Dis 1982;5:21-27.
- Hommes FA, Matsuo K. Effect of phenylalanine on brain maturation: Implications for the treatment of patients with phenylketonuria. In Wurtman RJ, Ritter-Walker E, eds. Dietary Phenylalanine and Brain Function. Boston: Birkhauser 1988, pp 238-243.
- Hommes FA, Moss L. Myelin turnover in hyperphenylalaninaemia. A re-evaluation with the HPH-5 mouse. J Inher Metab Dis 1992; 15:243-51.
- Hsaio KJ, Chen CH, Chiu FC, et al. A Chinese family with phenylketonuria and maternal phenylketonuria detected by family screening. Eur J Pediatr 1986;145:409-12.
- Huether G, Neuhoff V, Kaus R. Brain development in experimental hyperphenylalaninaemia: disturbed proliferation and reduced cell numbers in the cerebellum. Neuropediatrics 1983;14:12-19.
- Ikeda M, Levitt M, Udenfriend S. Phenylalanine as substrate and inhibitor of tyrosine hydroxylase. Arch Biochem Biophys 1967;120:420-427.
- Jablonska S, Stachow A, Suffczynska M. Skin and muscle indurations in phenylketonuria. Arch Dermatol. 1967;95:443-450.
- Johnson RC, Shah SN. Effect of hyperphenylalaninemia on fatty acid composition of lipids of rat brain myelin. J Neurochem 1973;21:1225-40.
- Kaufman S. An evaluation of the possible neurotoxicity of metabolites of phenylalanine. J Pediatr 1989:895-900.
- Kawashima H, Kawano M, Masaki A, Sato T. Three cases of untreated classical PKU: a report on cataracts and brain calcification. Am J Med Genet. 1988:29:89-93.
- Kayaalp E, Treacy E, Waters PJ, et al. Human phenylalanine hydroxylase mutations and hyperphenylalaninemia phenotypes: a metanalysis of genotype-phenotype correlations. Am J Hum Genet 1997;61:1309-1317.
- Kesby G. Repeated adverse fetal outcome in pregnancy complicated by uncontrolled maternal phenylketonuria. J Paediatr Child Health. 1999;35:499-502.
- Koch R, Fishler K, Azen C, et al. The relationship of genotype to phenotype in phenylalanine hydroxylase deficiency. Biochem Mol Med. 1997;60:92-101.
- Koepp P, Held KR. Serum-tyrosine in patients with hyperphenylalaninemia. Lancet 1977; 2:92.
- Kornreich HK, Shaw KNF, Koch R, Hanson V. Phenylketonuria and scleroderma. J. Pediatr. 1968;73:571-75.
- Krause W, Halminski M, McDonald L, Dembure P, Salvo R, Freides D, Elsas L. Biochemical and neuropsychological effects of elevated plasma phenylalanine in patients with treated phenylketonuria. A model for the study of phenylalanine and brain function in man. J. Clin Invest 1985 Jan; 75(1):40-8.
- Kreis R, Pietz J, Penzien J, et al. Identification and quantitation of phenylalanine in the brain of patients with phenylketonuria by means of in vivo 1H magnetic resonance spectroscopy. J Magn Reson B 1995:107:242-51.
- Lasser AE, Schultz BC, Beaff D, et al. Phenylketonuria and scleroderma. Arch Dermatol. 1978;114:1215-17.
- Ledley FD. Clinical application of genotypic diagnosis for phenylketonuria: theoretical considerations. Eur J Pediatr 1991;150:752-6.
- Ledley FD, Levy HL, Woo SLC. Molecular analysis of the inheritance of phenylketonuria and mild hyperphenylalaninemia in families with both disorders. N Engl J Med 1986;314:1276-80.
- Leuzzi V, Gualdi GF, Fabbrizi F, et al. Neuroradiological (MRI) abnormalities in phenylketonuric subjects: clinical and biochemical correlations. Neuropediatrics 1993;24:302-6.
- Leuzzi V, Trasimeni G, Gualdi GF, Antonozzi I. Biochemical, clinical and neuroradiological (MRI) correlations in late-detected individuals with PKU. J Inher Metab Dis 1995;18:624-34.
- Levy, HL. Phenylketonuria: old disease, new approach to treatment. Proc Nat Acad Sci USA 1999;96, 1811-1813.
- Lichter-Konecki U, Rupp A, Konecki DS et al. Relation between phenylalanine hydroxylase genotypes and phenotypic parameters of diagnosis and treatment of hyperphenylalaninemic disorders. J Inher Metab Dis. 1994;17:362-5.
- Ludolph AC, Vetter U, Ullrich K. Studies of multimodal evoked potentials in treated phenylketonuria: the pattern of vulnerability. Eur J Pediatr 1996 Jul;155 Suppl 1:S64-8.
- MacDonald A, Rylance GW, Asplin D, et al. Does a single plasma phenylalanine predict quality of control in phenylketonuria? Archives of Disease in Childhood, 1998;78,122-126.
- Mallolas J, Milà M, Lambruschini N, et al. Biochemical phenotype and its relationship with genotype in hyperphenylalaninemia heterozygotes. Molecular Genetics and Metabolism. 1999;67:156-61.
- Martynyuk AE, Savina S, Skibo GG. Blocking effect of intraperitoneal injection of phenylalanine on high-threshold calcium currents in rat hippocampal neurones. Brain Res 1991;552:228-231.
- Matsuo K, Hommes FA. The development of the muscarinic acetylcholine receptor in normal and hyperphenylalaninemic rat cerebrum. Neurochem Res 1988;13:867-70.
- Mazzocco MM, Yannicelli S, Nord AM, et al. Cognition and tyrosine supplementation among school-aged children with phenylketonuria. Am J Dis Child. 1992.146:1261-4.
- McCombe PA, McLaughlin DB, Chalk JB, et al. Spasticity and white matter abnormalities in adult phenylketonuria. Journal of Neurology, Neurosurgery and Psychiatry 1992;55:359-61.
- McDonald JD. The PKU mouse project: its history, potential and implications. Acta Paediatr 1994;407:122-3.
- McMurry MP, Chan GM, Leonard CO, Ernst SL. Bone mineral status in children with phenylketonuria--relationship to nutritional intake and phenylalanine control. Am J Clin Nurt 1992;55:97-1004.
- Melnick CR, Michals KK, Matalon R. Linguistic development of children with phenylketonuria and normal intelligence. J Pediatr. 1981;98:269-72.
- Michel U, Schmidt E, Batzler U. Results of psychological testing of patients aged 3-6 years. Eur J Pediatr. 1990;149 Suppl 1:S34-8.
- Möller HE, Weglage J, Wiedermann D, Ullrich K. Blood-brain barrier phenylalanine transport and individual vulnerability in phenylketonuria. J Cereb Blood Floow Metab 1998; 18:1184-91.
- Mostafawy A, Nagle JB, Newill A, Braatz E. A study of cerebral atrophy in phenylketonuria. Neuropädiatrie. 1970;2:215-25.
- Newborn Screening Committee, The Council of Regional Networks for Genetics Services (CORN), National Newborn Screening Report – 1994, CORN, Atlanta, January 1999.
- Nishimura N, Okamoto H, Yasui M, et al. Intermediary metabolism of phenylalanine and tyrosine in diffuse collagen diseases II. Influences of the low phenylalanine and tyrosine diet upon patients with collagen diseases. Arch Dermatol. 1959; 80:466-76.
- Nova MP, Kaufman M, Halperin A. Scleroderma-like skin indurations in a child with phenylketonuria: A clinicopathologic correlation and review of the literature. J Am Acad Dermatol. 1992;26:329-33.
- Novotny EJ, Avison MJ, Herschkowitz N, et al. In vivo measurement of phenylalanine in human brain by proton nuclear magnetic resonance spectroscopy. Pediatr Res 1995;37:244-49.
- OkanoY, Asada M, Kang Y, et al. Molecular characterization of phenylketonuria in Japanese patients. Hum Genet 1998; 103:613-8.
- Okano Y, Eisensmith RC, Güttler F, et al. Molecular basis of phenotypic heterogeneity in phenylketonuria. N Engl J Med. 1991;324:1232-8.
- Online Mendelian Inheritance in Man. OMIM (TH). Center for Medical Genetics, Johns Hopkins University (Baltimore, MD), and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD). URL: http://www.ncbi.nlm.nih.gov/omim.
- Özalp I, Coskun T, Özgüc M, et al. Genetic and neurological evaluation of untreated and late-treated patients with phenylketonuria. J Inher Metab Dis. 1994;17:371.
- Ozanne AE, Krimmer H, Murdoch BE. Speech and language skills in children with early treated phenylketonuria. Am J Mental Retard. 1990;94:625-32.
- Pardridge WM. Kinetics of competitive inhibition of neutral amino acids across the blood-brain barrier. J Neurochem 1977;29:103-8.
- Patel MS, Arinze IJ. Phenylketonuria: metabolic alterations induced by phenylalanine and phenylpyruvate. Am J Clin Nutr 1975; 28:183-188.
- Pearsen KD, Gean-Marton AD, Levy HL, Davis KR. Phenylketonuria: MR imaging of the brain with clinical correlation. Radiology. 1990;177:437-40.
- Pennington BF, Smith SD. Genetic influences on learning disabilities and speech and language disorders. Child Development. 1983;54:369-87.
- Pérez B, Desviat LR, Garcia MJ, Ugarte M. Different phenotypic manifestations associated with identical phenylketonuria genotypes in two Spanish families. J Inher Metab Dis. 1994;17:377-8.
- Perry T, Hansen S, Tischler B, et al. Unrecognized adult phenylketonuria: implications for obstetrics and psychiatry. N Engl J Med 1973;289:395-8.
- Pietz, J. Neurological aspects of adult phenylketonuria. Current Opinion in Neurology 1998; 11: 679-688.
- Pietz J, Benninger C, Schmidt H, et al. Long-term development of intelligence (IQ) and EEG in 34 children with phenylketonuria treated early. Eur J Pediatr. 1988; 147:361-7.
- Pietz J, Dunckelmann R, Rupp A, et al. Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr. 1998;157:824-30.
- Pietz J, Fatkenheuer B, Burgard P, et al. Psychiatric disorders in adult patients with early-treated phenylketonuria. Pediatrics. 1997;99:345-50.
- Pietz J, Kreis R, Boesch C, et al. The dynamics of brain concentrations of phenylalanine and its clinical significance in patients with phenylketonuria determined by in vivo 1H magnetic resonance spectroscopy. Pediatr Res 1995:38:657-663.
- Pitt D. Phenylketonuria with normal intelligence: report of two cases. Aust J Ment Res. 1971;1:160-3. In Tyfield, Zachocke et al. 1995.
- Pitt DB, O'Day J. Phenylketonuria does not cause cataracts. Eur J Pediatr 1991; 150:661-4.
- Popescu A, Andrian T, Güttler F, Guldberg P. Genotype-phenotype correlation in 11 Romanian PKU families. J Inher Metab Dis. 1994;17:374-5.
- Poser CM, Van Bogaert L. Neuropathologic observations in phenylketonuria. Brain 1959;82:1-8.
- Primrose DA. Phenylketonuria with normal intelligence. J Ment Def Res. 1983;27:239-246.
- Ramus SJ, Forrest SM, Pitt DD, Cotton RGH. Genotype and intellectual phenotype in untreated phenylketonuria patients. Pediatr Res. 1999;45:474-81.
- Ramus S, Forrest SM, Pitt D, et al. Comparison of genotype and intellectual phenotype in untreated individuals with PKU. J Med Genet. 1993;30:401-5.
- Romeo G, Menozzi P, Ferlini A, et al. Incidence of classic PKU in Italy estimated from consanguineous marriages and from neonatal screening. Clin Genet 1983; 24:339-345.
- Saugstad LF. Frequency of phenylketonuria in Norway. Clin Genet 1975;7:40-51.
- Schmidt E, Rupp A, Burgard P, Pietz J, Weglage J, de Sonneville L. Sustained attention in adult phenylketonuria: the influence of the concurrent phenylalanine-blood-level. J Clin Exp Neuropsychol 1994 Oct;16(5):681-8.
- Scriver CR, Eisensmith RC, Woo SLC, Kaufman S. The hyperphenylalaninemias of man and mouse. Ann Rev Genet. 1994;28(4):141-65.
- Scriver, CR, Kaufman S, Eisensmith RC, Woo SLC. (1995). The hyperphenylalaninemias. In: The metabolic basis of inherited disease, vol. 1, Scriver, C. R., Beaudet, A. L., Sly, W. S., & Valle, D. (Eds)., New York, McGraw Hill, (7th ed), pp. 1015-1075.
- Scriver CR., Waters PJ. (1999). Monogenic traits are not simple. Lessons from phenylketonuria. Trends in Genetics, 15(7), 267-272.
- Shah SN. Fatty acid composition of lipids of human brain myelin and synaptosomes: changes in phenylketonuria and Down's syndrome. Int J Biochem 1979; 10:477-82.
- Shaw DW, Maravilla KR, Weinberger E, et al. MR imaging of phenylketonuria. Am J Neuroradiol 1991;12:403-6.
- Shaw DWW, Weinberger E, Maravilla KR. Cranial MR in phenylketonuria. Journal of Computer Assisted Tomography. 1990; 14:458-60.
- Stemerdink BA, van der Meere JJ, van der Molen MW, et al. Information processing in patients with early and continuously-treated phenylketonuria. Eur J Pediatr 1995;154:739-46.
- Superti-Furga A, Steinmann B, Duc G, Gitzelmann R. Maternal phenylketonuria syndrome in cousins caused by mild, unrecognized phenylketonuria in their mothers homozygous for the phenylalanine hydroxylase Arg-261-Gln mutation. Eur J Pediatr 1991;150:493-7.
- Svensson E, von Döblein U, Eisensmith RC, et al. Relation between genotype and phenotype in Swedish phenylketonuria and hyperphenylalaninemia patients. Eur J Peidatr 1993;152:132-9.
- Svensson E, von Döblein U, Hagenfeldt L. Polymorphic DNA haplotypes at the phenylalanine hydroxylase locus and their relation to phenotype in Swedish phenylketonuria families. Hum Genet. 1991; 87:11-17.
- Teebi AS, Al-Awadi SA, Farag TI, et al. Phenylketonuria in Kuwait and Arab countries. Eur J Pediatr 1987;146:59-60.
- Thalhammer O. Frequency of inborn errors of metabolism, especially PKU, in some representative newborn screening centers around the world. Hum Genet 1975;30:273-286.
- Thompson AJ, Smith I, Brenton D, et al. Neurological deterioration in young adults with phenylketonuria. Lancet 1990;336:602-605.
- Thompson AJ, Smith I, Kendall BE, et al. Magnetic resonance imaging changes in early treated patients with phenylketonuria. Lancet 1991;337:1224.
- Toft PB, Lou HC, Krägeloh-Mann I, et al. Brain magnetic resonance imaging in children with optimally controlled hyperphenylalaninemia. J Inher Metab Dis 1994;17:575-583.
- Treacy E, Pitt JJ, Seller K, Thompson GN, Ramus S, Cotton RG. In vivo disposal of phenylalanine in phenylketonuria: a study of two siblings. J Inherit Metab Dis 1996;19(5):595-602.
- Trefz FK, Burgard P, König T, Goebel-Schreiner B, et al. Genotype-phenotype correlations in phenylketonuria. Clin Chim Acta. 1993;217:15-21.
- Tyfield, LA. Phenylketonuria in Britain: genetic analysis gives a historical perspective of the disorder but will it predict the future for affected individuals? J Clin Pathol: Mol Pathol 1997;50, 169-174.
- Tyfield LA, Meredith AL, Osborn MJ, et al. Genetic analysis of treated and untreated phenylketonuria in one family. J Med Genet. 1990;27:564-8.
- Tyfield LA, Zschocke J, Stephenson A, et al. Discordant phenylketonuria phenotypes in one family: the relationship between genotype and clinical outcome is a function of multiple effects. J Med Genet 1995;32:867-70.
- Ullrich K, Möller H, Weglage J, et al. White matter abnormalities in phenylketonuria: results of magnetic resonance measurements. Acta Paediatr Suppl 1994;407:78-82.
- Ullrich K, Weglage J, Oberwittler C, et al. Effect of L-DOPA on visual evoked potentials and neuropsychological tests in adult phenylketonuria patients. Eur J Pediatr 1996;155(Suppl 1):S74-S77.
- Usha R, Uma R, Farag TI, et al. Late diagnosis of phenylketonuria in a Bedouin mother. Am J Med Genet. 1992;44:713-5.
- Verkerk PH, van der Meer SB. Failure to detect phenylketonuria. Eur J Pediatr. 1995;154:163.
- Verkerk PH, van Spronsen FJ, Smit GP, et al. Prevalence of congenital heart disease in patients with phenylketonuria. J Pediatr. 1991;119:282-283.
- Villasana D, Butler IJ, Williams JC, Roongta SM. Neurological deterioration in adult phenylketonuria. J Inher Metab Dis 1989;12:451-457.
- Waisbren SE, Levy HL. Agoraphobia in phenylketonuria. J Inher Metabol Dis. 1991;14:755.
- Wall KM, Pardridge WM. Decreases in brain protein synthesis elicited by moderate increases in plasma phenylalanine. Biochem Biophys Res Comm 1990; 168:1177-83.
- Weglage J, Bick U, Schuierer G, et al. Progression of cerebral white matter abnormalities in early treated patients with phenylketonuria during adolescence. Neuropediatrics 1997;28:239-40.
- Weglage J, Fünders B, Ullrich K, et al. Psychosocial aspects in phenylketonuria. Eur J Pediatr. 1996;155Suppl1:S101-4.
- Weglage J, Fünders B, Wilken B, Schubert D, Schmidt E, Burgard P, Ullrich K. Psychological and social findings in adolescents with phenylketonuria. Eur J Pediatr 1992 Jul;151(7):522-5
- Weglage J, Schuierer G, Kurlemann G, et al. Different degrees of white matter abnormalities in untreated phenylketonurics: findings in magnetic resonance imaging. J Inher Metab Dis 1993;16:1047-8.
- Weglage, J., Ullrich, K., Pietsch, M., et al. Intellectual, neurologic, and neuropsychologic outcome in untreated subjects with nonphenylketonuria hyperphenylalaninemia. Pediatric Research 1997;42:378-384.
- Welsh MC, Pennington BF, Ozonoff S, et al. Neuropsychology of early-treated phenylketonuria: specific executive function deficits. Child Dev. 1990;61:1697-1713.
- Wood B. Neurological disturbance in a phenylketonuric child after discontinuance of dietary treatment. Develop Med Child Neurol. 1976;18:657-65.
- Woodring JH, Rosenbaum HD. Bone changes in phenylketonuria reassessed. Am J Roentgenol 1981;137:241-3
- Zartler AS, Sassaman E. Linguistic development in PKU [letter]. J. Pediatr 1981 Sep;99(3):501.
 BACK TO TOP
BACK TO TOP